
Chromatographic, Spectrometric and NMR Characterization of a New Set of Glucuronic Acid Esters Synthesized by Lipase
Unit of General and Organic Chemistry. Gembloux Agricultural University. Passage des Déportés, 2. B-5030 Gembloux (Belgium). E-Mail: lipazben@hotmail.com
Unit of Analytical Chemistry. Gembloux Agricultural University. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
Unit of Food Technology. Gembloux Agricultural University. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
Unit of Bio-industry. Gembloux Agricultural University. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
Laboratory of Organic Chemistry. University of Mons-Hainaut. Avenue Maistriau, 19. B-7000 Mons (Belgium).
University of Orléans. ICOA-UMR 6005. BP 6759, F-45067 Orléans Cedex 2 (France).
Unit of Biological Chemistry. Gembloux Agricultural University. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
Unit of General and Organic Chemistry. Gembloux Agricultural University. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
Received on 10 February 2006, accepted on 26 September 2006.
Résumé
Caractérisation chromatographique, spectrométrique et RMN d'une nouvelle série d'esters d'acide D-glucuronique synthétisée à l'aide d'une lipase.La synthèse enzymatique (lipase de Candida antarctica) a été développée afin de synthétiser une série de nouveaux esters à partir de l'acide D-glucuronique. Les identifications par TOF/MS et MS/MS confirment la synthèse de ces esters d'acide D-glucuronique. Les analyses chromatographiques ont révélé la présence de formes anomériques pour les esters de longueurs de chaîne carbonée supérieures à 12. L'étude par RMN confirme la structure des différents esters d'acide D-glucuronique et met en évidence l'existence des formes anomériques.
Abstract
An enzymatic synthesis was developed on a new set of D-glucuronic acid esters and particularly the tetradecyl-D-glucopyranosiduronate also named tetradecyl D-glucuronate. Chromatographic analyses revealed the presence of the ester as a mixture of anomeric forms for carbon chain lengths superior to 12. TOF/MS and MS/MS studies confirmed the synthesis of glucuronic acid ester. The NMR study also confirmed the structure of glucuronic acid esters and clearly revealed an anomeric (α/β) ratio equivalent to 3/2.
1. Introduction
1Sugar-derived fatty esters, which are non-ionic surfactants, have good surface properties, and total biodegradability. Their tensio-active properties result from the occurrence of two complementary moieties on the same molecule. Due to their tastelessness, non skin-irritation and non-toxicity, they find diverse uses in the cosmetic, food and pharmaceutical industries (Picciuto et al., 2001). The application of standard chemical procedures for the synthesis of sugar-derived fatty esters requires high energy consumption and usually results in the formation of a mixture of compounds (due to the presence of primary and secondary sugar hydroxyl groups) and sometimes of unwanted colored side-products (Sarney,Vulfson, 1995; Vulfson, 1998). Enzymatic synthesis has long been considered as an alternative methodology for the elaboration of sugar-derived esters and, nowadays, this way of preparation is largely documented (Cao et al., 1996; Basso et al., 2002).
2In vivo, D-glucuronic acid esters are found in urine, where they are involved in the conversion of arylalkanoic acid. They are also partly used to target antitumor drugs like Paclitaxel (De Bont et al., 1997) or like prodrug selectively activated in tumors by extracellular human β-glucuronidase (De Graaf et al., 2004). Moreover, these syntheses thanks to lipases are an alternative to the synthesis of unsaturated glucose esters of pharmaceutical interest (Otto et al., 1998; Otto et al., 2000).
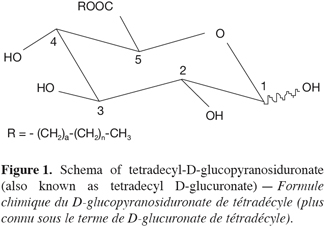
3The results, reported herein for the first time, focus on production in 2 l reactor of a set of D-glucuronic acid esters using Candida antarctica B lipase (lipase: triacylglycerol hydrolases, EC 3.1.1.3) and on the identification of these compounds, which are an unusual non-ionic surfactant namely alkyl-D-glucopyranosiduronate (also known as alkyl D-glucuronate) (Figure 1). These results are the supplementary to those experimented in flasks and earlier published (Moreau et al., 2004). These glucuronic acid esters should certainly have quite similar properties when compared to the glucose esters. The esters were highly purified in order to investigate in details their surface properties, and preliminary investigations had demonstrated relevant results for the C8 to C14 homologous (Blecker et al., 2002). Both yield and purity of each ester were checked by RP-HPLC with evaporative light scattering detection. The novel glucuronic acid esters have been fully characterized by GC-MS and TOF-MS, ionspray® mass spectrometry, FT-IR and 1H- and 13C-NMR.
2. Experiment and Reagents
4Immobilized lipase B from Candida antarctica (Novozym SP435) was obtained from Novo Nordisk. Pure D-glucuronic acid and 0.4 nm molecular sieves (8-12 mesh) were purchased from Sigma. Primary alcohols with chain length ranging from C8 to C16 (all above 99% purity) were purchased from Merck. Analytical grade tert-butyl alcohol was purchased from Fluka. All the solvents used in the extraction procedure and in chromatography were of HPLC grade.
2.1. Enzymatic synthesis of sugar-derived fatty esters
5Tert-butyl alcohol solutions (500 ml) containing 100 mmol of reagents (either D-glucuronic acid and alcohol or glucose and fatty acid) were added to a 2 l reactor heated at 60°C. Molecular sieve (0.4 nm, 25 g) was added to remove water produced during the reaction. The reactions were carried out at 60°C during 48 h, under 150 rpm and started with adding of 5 g of Candida antarctica lipase. The amount of lipase and the molar ratio of reagent were studied to optimize the esterification yield. At the end of the reaction, the lipase and molecular sieves were removed by filtration.
2.2. Purification of the esters
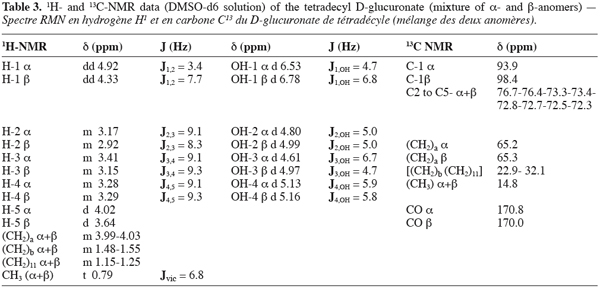
6The filtrate containing the products was diluted with 350 ml distilled water and the mixture was mixed at 150 rpm during 2 min. After that, 2 portions of 400 ml dichloromethane were introduced in the reactor in keeping the agitation during 10 min. The raw dichloromethane extracts were pooled and the solvent was evaporated to dryness under reduced pressure at 35°C. The solid residue was purified by adsorption chromatography using 300 mm x 60 mm I.D. glass columns packed in pure chloroform (stabilized with 0.5% ethanol) with 70-230 mesh G60 silicagel previously conditioned at a 3.5% water content. After elution of the residual free alcohol in two steps with 100 ml of chloroform/methanol (90:10, V:V) followed by 100 ml of chloroform/methanol (80:20, V:V), the molecules of interest were recovered by elution with a chloroform/methanol (70:30, V:V) mixture. Two ml fractions were collected and concentrated under a gentle stream of nitrogen. The occurrence of ester was systematically checked on each fraction by thin layer chromatography on Si-60 (80 x 40 mm) plates from Macherey-Nagel with chloroform/methanol/water (65:15:2, V:V) as mobile phase. The spots were visualized after spraying with a reagent containing acetic acid/sulfuric acid/anisaldehyde (100:2:1, V:V) and then followed by heating at 150°C for 10 min. Depending on their chain length, the Rf values of the different esters ranged from 0.42 to 0.58. The fractions containing only the interesting molecules were pooled and the solvent evaporated under reduced pressure at 35°C. In such a way, pure compounds (> 98%) could easily be recovered.
2.3. Chemical analysis and characterization of the synthesized esters
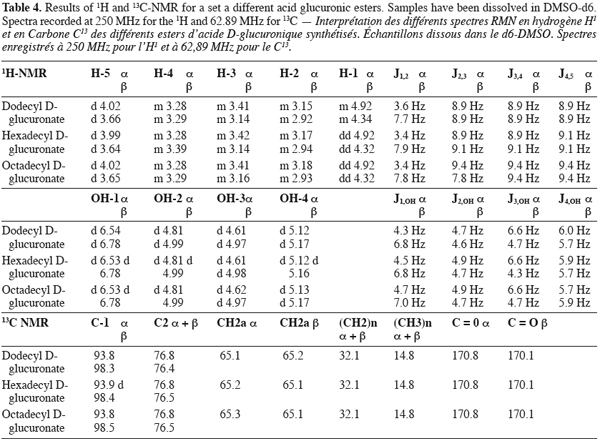
7The different molecules were checked for purity by RP-HPLC on a Hewlett-Packard HP1050 chromatograph coupled with an evaporative light scattering detector (Alltech model 500 ELSD) with a drift tube maintained at 84.5°C and a nebulizer gas flow rate of 2.22 standard liter per minute. The analyses were performed on a RP-18 end capped Inertsil 5 ODS2, 250 x 2 mm I.D. column (from Varian). The elutions were carried out in the isocratic mode with acetonitrile/water (65:35, V:V) as mobile phase with a flow rate fixed at 0.3 ml/min.
8All new glucuronates were fully characterized by a series of spectrometric investigations: NMR spectra were recorded on a Bruker Avance DPX 250 (250 MHz for the 1H-NMR and 68.89 MHz for the 13C-NMR) as d6-Me2SO solutions. Ionspray® spectra were recorded on a Perkin Elmer SCIEX API 300 MS (the samples were solubilized in acetonitrile/methanol 90:10, V:V and FT-IR spectra were recorded on a Bruker IFS 48 spectrometer. ESI-TOF-MS and ESI-TOF-MS/MS spectra were recorded on a Waters Q-TOF-2 mass spectrometer, the samples (10-4 M) were solubilized in acetonitrile/methanol 90:10, V:V in which an equal volume of NaI (10-4 M) was added. The solutions were infused into the ESI source at a rate of 5 l.min-1 with a Harvard syringe pump. Typical ESI conditions were: capillary voltage: 3.1 kV ; cone voltage: 80 V; source temperature: 80°C and desolvation temperature: 120°C. Dry nitrogen was used as the ESI gas. The quadrupole was set to pass ions from 100 to 3000 Th, and all ions were transmitted into the pusher region of the time-of-flight analyzer, where they were mass-analyzed with a 1 sec integration time. Data were acquired in continuum mode until acceptable averaged data were obtained. The MS/MS experiments were performed by mass-selecting the parent ion beam with the quadrupole (Q) and acceleration to 25 eV kinetic energy. The ions were then subjected to collisions with Argon in the hexapole cell and the fragments were mass-measured by the TOF analyzer (TOF).
3. Results and discussion
3.1. Esterification of esters using Candida antarctica lipase
9As previously described (Moreau et al., 2004), direct enzymatic esterification of a set of alkyl branched D-glucuronic acid esters was synthesized in flasks for the first time. Consequently, the synthesis of D-glucuronic acid esters was started in a 2 l reactor and was compared to the synthesis of glucose esters in the same reaction conditions. The table 1 showed that the reaction on a large scale increases the molar yield of synthesis of both kinds of esters. This increasing in yield compared with those recorded in flasks could be explained by a better homogeneity inside the media in the reactor.
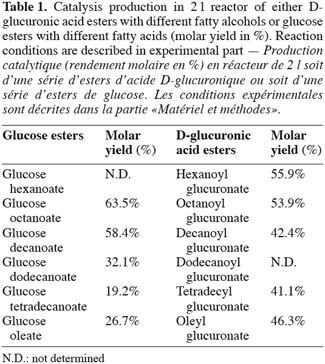
10At the end of those experiments carried out in reactor, the influence of the chain length on molar yield was more marked in the case of glucose ester synthesis than in those concerning the D-glucuronic acid esters. Actually, we could explain these results on the basis of the structure of the acyl donor (D-glucuronic acid). It was well-known that during an enzymatic esterification, the acyl donor enters in first in the catalytic site of the lipase (Garcia et al., 1999). In the case of esterification of uronic esters, the acyl donor is more bulky than a fatty acid (acyl donor for the esterification of a sugar ester). As a result the length of alcohol exerts less influence during an esterification of uronic acid. This fact was confirmed by the enzymatic esterification of glucose. In this latter case, the molar yield decreased with the acyl donor chain length and proved also the specificity of Candida antarctica lipase for short or medium acyl chain, corroborating thereby the theoretical studies of Pleiss et al. (1998) and Schmidt-Dannert (1999). These results tended to demonstrate that the inversion of position acyl moiety has a repercussion on the reaction yield. All these compounds were very pure (more than 98% by RP-HPLC).
11The optimization of the esterification process in 2 l reactor focused on two parameters (amount of lipase and molar ratio of reagents). The increasing lipase amount promoted the synthesis of esters. For large-scale applications, where the price of lipase has to be taken into account, these experiments revealed that 2.5 g of enzyme were enough to get a good yield (Table 2).

12When synthesizing glucose esters, the optimization of molar ratio (mol acyl donor/mol acyl acquirement) proved that an excess of acyl donor gave better results than an excess of acyl acquirement (Figure 2). These results supported different studies (Ljunger et al., 1994; Coulon et al., 1996; Tsitsimpikou et al., 1997). Conversely, the excess of glucose could lead to a distortion between the enzyme-water interaction and, in the worst case, it could strip water off the enzyme. The explanation for the increase in the molar yield could also be found in the reactive mechanism. This lipase is known as the enzyme for bulkier substrate; the increase in oleic acid could promoted the conversion of «enzyme» into the binary complex «enzyme-oleic acid» and release of water, which would be followed by the other binary complex «Acyl-enzyme-glucose». In other words, the increase in molar yield shifted the equilibrium to the synthesis of ester. On the contrary, the increase in glucose inhibited the synthesis of ester. This observation backs the study of Arcos et al. (2001), which confirmed the esterification of glucose with a fatty acid as a «Ping-pong bi-bi» kinetic.
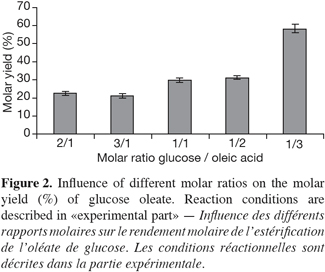
13Higher concentrations of acyl donor were not tested in order to avoid a more important accumulation of fatty acid in the media which could load down the purification steps.
14In the case of D-glucuronic acid esters, an excess of acyl donor was not considered because D-glucuronic acid was bulkier, and therefore the enzyme would be saturated faster.
3.2. Identification of a new set of D-glucuronic acid esters
15After the catalytic reaction (Novozym SP 435 lipase) between D-glucuronic acid and tetradecanol in a thermostatised 2 l reactor, a TLC analysis of the recovered molecules revealed a double spot at Rf 0.5 and 0.6; the spot at Rf 0.85 was identified as residual alcohol. Figure 3 shows a typical RP-HPLC chromatogram recorded for the purified 1-tetradecyl D-glucuronate. Beside minute amounts of residual long chain alcohol, two peaks at Rt 10.0 and 11.0 min were visualized. These peaks were tentatively attributed to the glucuronates anomeric forms.
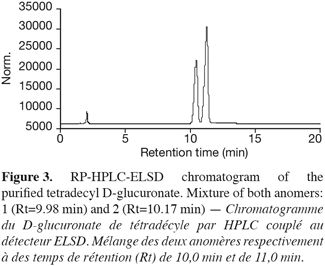
16Nevertheless, at this point of the work, we cannot say at which peak which α or β anomeric form corresponds. Chromatography on a graphite column could provide some information to solve this problem.
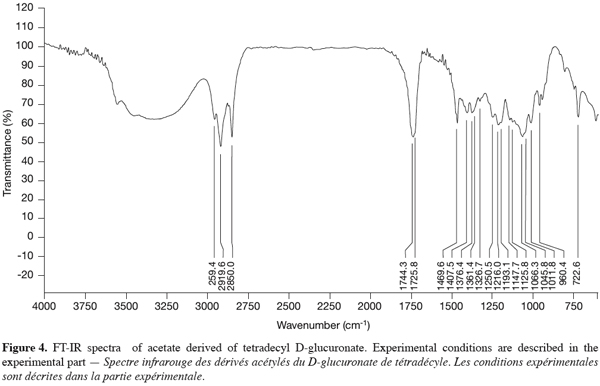
17The FT-IR spectra revealed two characteristic bands at 3420-3500 cm-1 and 1744 cm-1, respectively attributed to the OH bands and the ester linkage (Figure 4), also described by Seino et al. (1984). The other bands were attributed to the alkyl moieties of the molecule.
18The GC-MS chromatogram of 1-tetradecyl D-glucuronate, derived as the acetate esters as far as the hydroxyl groups are concerned features two peaks (at Rt 43.8 and 44.3 min) which indeed corroborate the peaks seen in reverse phase HPLC. These two peaks represent an additional clue of the presence of anomeric forms. Nevertheless, given the fact that the mass spectrometer works in the electron ionization mode that is recognized as a hard ionization mode, the recorded mass spectra do not present any signal corresponding to the molecular ions of the derived carbohydrates and only fragments ions are observed. Consequently, this experiment does not supply an unambiguous proof attesting the successful catalytic synthesis of the compounds.
19To overcome this problem, it is often required, especially in the context of biomolecules, to use a softer ionization mode, such as electrospray ionization (ESI). In the ESI spectrum recorded on an Ionspray® mass spectrometer, the occurrence of adduct ions at m/z 408 (M+NH4)+; m/z 413 (M+Na)+ and m/z 429 (M+K)+ was in keeping with the ionization of a molecule of 390 Daltons, the molecular mass of 1-tetradecyl D-glucuronate. In the same way, all the prepared alkyl glucuronates (C8 to C16) presented under ESI ionization the same behavior, confirming their successful preparation under the conditions reported here above.
20In order to obtain more information on the structure of the obtained molecules, we decided to perform some additional experiments on the Waters Q-TOF-2 tandem mass spectrometer that allows the recording of MS-MS experiments on ESI prepared ions. The selected target molecule was again the 1-tetradecyl D-glucuronate. First of all, the TOF-MS spectrum showed the specific mass-to-charge ratio at m/z 413 expected on the basis of a Na+ complexation (M+Na)+ (Figure 5).
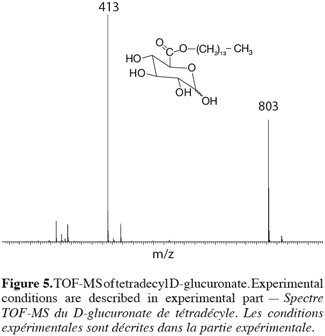
21In addition to this signal, a peak at m/z 803 was also recorded and corresponded to a complex ion between two ester molecules and a sodium ion (2M+Na)+. The comparison between the experimental isotopic pattern and a theoretical simulation of the isotopic ions distribution revealed a close similarity that readily confirms the elemental composition of the ionic species. More details into the structure of the m/z 413 ions can, as introduced here above, be obtained by tandem mass spectrometry (TOF-MS/MS), especially because glucose myristate was available as a referent isomeric species (Figure 6).
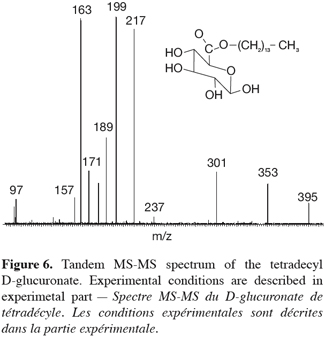
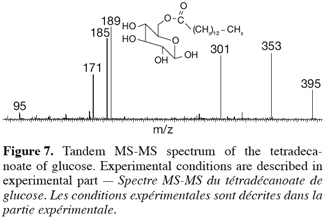
22Both species are obtained in the gas phase of the mass spectrometer as [M+Na]+ species upon ESI ionization. Their MS-MS spectra were compared in figure 7. First of all, both ions present different fragmentations upon collision-induced dissociation and that was of course expected when dealing with isomeric ions. The MS-MS spectrum of m/z 413 ions coming from the 1-tetradecyl D-glucuronate molecule showed inter alia interesting signals at m/z 395, 217 and 199, respectively corresponding to the losses of H2O, C14H28 and C14H29OH. In particular, the two latter fragmentations were in agreement with the presence of the –O-(CH2)13-CH3 group. In the MS/MS spectrum of the referent ions, the corresponding signals have totally disappeared and a structural informative signal occurs at m/z 185. This peak corresponded to ions coming from the collision-induced loss of myristic acid, CH3-(CH2)12COOH, expected on the basis of the starting molecules, see figure 7. This comparison between both the MS/MS spectra unambiguously confirmed the structure of the enzymatic prepared ester, namely 1-tetradecyl D-glucuronate. A mechanistic study of the observed fragmentations is currently undertaken and will be published separately.
23The 1H and 13C-NMR patterns, gathered in table 3, confirmed final structure proof of synthesis of the 1-tetradecyl D-glucuronate. The values, recorded in 1H NMR, of 3.99-4.03 ppm corresponding to (CH2) in position «a» of the ester linkage (as described in figure 1) were markedly more important than values for the other CH2 on the length alkyl chain (more important unshielding than the other CH2). Except the values for H-1, high values of H-5, compared to H-2,-3 and-4, and interpretation of results recorded in 13C NMR were much as proofs of the synthesis of a D-glucuronic acid ester. It was very interesting to confirm the presence of anomeric forms of compound synthesized. In fact, the value of H-1 α (4.92 ppm) was superior than value of H-1 β (4.33 ppm), corroborating the dogma for which the equatorial position of hydrogens are always more unshielded than those in axial position. In addition, those values were higher than other hydrogen values in position 2, 3, 4 and 5. An other argument for the presence of anomeric forms was obtained from the vicinal coupling constants. In fact, J1,2 (7.7Hz) of H-1 β was more important than the other in H-1 α (3.4Hz). All these results confirmed the presence of anomeric forms of 1-tetradecyl D-glucuronate. In addition, the 1H NMR experiments assessed the anomeric ratio (α/β) at 3/2. The results in 1H and 13C recorded were in line with those of Ramos et al. (1996) or Wessel and Bartsch (1995). It was also interesting to note the value of J1,2 of H-1 β shape. This value (7.7Hz) for a D-glucuronic acid ester was always smaller than the value of J1,2 of D-glucuronic acid (9-10Hz). The reason of this observation could be attributed to a deformation of the pyrano ring witch was usually in the 4 C1 conformation.
24A set of syntheses of D-glucuronic acid esters with alcohol chain length was undertaken and led to the production of very pure molecules to C8 to C16. The increase of chain length could have influence on HLB balance and thus on chemical and biological properties.
25The 1H and 13C NMR results gathered in table 4 allowed to confirm the synthesis of a set of D-acid glucuronic esters. Moreover, they also confirmed the existence of anomeric forms of those compounds.
26Carbohydrate fatty esters are very attractive surfactants because various tensio-active properties can be obtained depending on their structure. Concerning the n-alkyl esters of D-glucuronic acid, it was established that these molecules can present interesting properties depending on the hydrophobic chain length (Blecker et al., 2002). For example, the important adsorption speed of decyl glucuronate and dodecyl glucuronate allowed them to cause a significant decrease of surface tension in a few seconds. This characteristic could be associated to foaming properties. Micellar structures could be obtained using a relatively small bulk concentration of octyl glucuronate. So the use of this molecule could be envisaged in detergents or generally speaking, in systems where a hydrophobic compound must be dispersed in an aqueous phase. Such systems are found in drugs and cosmetic formulations.
4. Conclusion
27In this paper, a set of different D-glucuronic acid esters (C8 to C18:1) was synthesized for the first time in 2 l reactor allowing yield improvement during esterification in comparison with earlier study (Moreau et al., 2004). In addition, the yields of this set of glucuronic acid esters were equivalent explaining in this way that the D-glucuronic acid (acyl donor) was bulkier than a traditional acyl donor like fatty acid and in consequence the inversion of acyl moiety position had an impact on the reaction yield.
28This paper highlighted the identification by different chromatographic and spectrometric techniques of a new set of D-glucuronic acid esters. The RP-HPLC analyses led to the separation of both α and β anomeric forms of these compounds. Moreover, longer was the alkyl chain, better was the chromatographic resolution. Tandem MS/MS confirmed the synthesis of tetradecyl D-glucuronate that was also attested by NMR results. An anomeric ratio (α/β) of 3/2 was established by 1H-NMR.
29These compounds, prepared by a lipase, represent new various biosurfactants and eventually more compounds could be obtained by adapting the length of the hydrophobic chain linked to the D-glucuronic acid moiety. The detailed study of both surface properties and functionalities of the new molecules is in progress.
Acknowledgements
30The interdisciplinary team associated to the study wish to acknowledge the “Ministère de la Communauté française” for financial support and thanks the “Fonds National pour la Recherche Scientifique” for financial support in the acquisition of the Waters Q-TOF-2 instrument and for continuing support (Research associate).
Abreviations
31ELSD: Evaporatif light scattering diffraction;
32HPLC: High pressure liquid chromatography;
33RP-HPLC: Reverse phase-high pressure liquid chromatography;
34NMR: Nuclear magnetic resonance;
35ESI/TOF/MS: Electro spray ionization/Time of fly/Mass spectrometry;
36FT-IR: Fourier transformation-Infra red;
37TLC: Thin liquid chromatography;
38GC-MS: Gas chromatography-mass spectrometry.
Bibliographie
Arcos JA., Hill CG., Otero C. (2001). Kinetics of the lipase-catalyzed synthesis of glucose esters in acetone. Biotechnol. Bioeng. 73 (2), p. 104–110.
Basso A., Ducret A., Gardossi L., Lortie R. (2002). Synthesis of octyl glucopyranoside by almond β-glucosidase adsorbed onto celite R-640. Tetrahedron Lett. 43, p. 2005–2008.
Blecker C., Piccicuto S., Lognay G., Deroanne C., Marlier M., Paquot M. (2002). Enzymatically prepared n-alkyl esters of glucuronic acid: the effect of hydrophobic chain length on surface properties. J. Coll. Interf. Sci. 247, p. 424–428.
Cao L., Fischer A., Bornscheuer UT., Schmid RD. (1996). Lipase-catalyzed solid phase synthesis of sugar fatty acid esters. Biocatal. Biotransform. 14, p. 269–283.
Coulon D., Ismail A., Girardin M., Rovel B., Ghoul M. (1996). Effect of different biochemical parameters on the enzymatic synthesis of fructose oleate. J. Biotechnol. 51, p. 115–121.
De Bont D., Leenders R., Haisma H., Van der Meulen-Muileman I., Scheeren HW. (1997). Synthesis and biological activity of beta-glucuronyl carbamate based produrugs of paclitaxel as potential candidates for ADEPT. Bioorg. Med. Chem. 5, p. 405–414.
De Graaf M., Nevalainen T., Scheeren H., Pinedo H., Haisma H., Boven E. (2004). A methyl ester of the glucuronide prodrug DOX-GA3 for improvement of tumor-selective chemotherapy. Biochem. Pharmacol. 68, p. 2273–2281.
Garcia T., Sanchez N., Martinez M., Aracil J. (1999). Enzymatic synthesis of fatty esters. Part II. Kinetic approach. Enzyme Microb. Technol. 25, p. 584–590.
Ljunger G., Aldercreutz P, Mattiasson B (1994). Lipase catalysed acylation of glucose. Biotechnol. Lett. 16, p. 1167–1172.
Moreau B., Lognay GC., Blecker C., Brohée JC., Chéry F., Rollin P., Paquot M., Marlier M. (2004). Synthesis of novel D-glucuronic acid fatty esters using Candida antarctica lipase in tert-butanol. Biotechnol. Lett. 26, p. 419–424.
Otto RT., Bornscheuer UT., Scheib H., Pleiss J., Syldatk C., Schmid RD. (1998). Lipase catalysed esterification of unsual substrates : synthesis of glucuronic acid and ascorbic acid (vitamin C) esters. Biotechnol. Lett. 20, p. 1091–1094.
Otto RT., Scheib H., Bornscheuer UT., Pleiss J., Syldatk C., Schmid RD. (2000). Substrate specificity of lipase B from Candida antarctica in the synthesis of arylaliphatic glycolipids. J. Mol. Catal. Part B. Enzym. 8, p. 201–211.
Picciuto S., Blecker C., Brohée JC., Mbampara A., Lognay G., Deroanne C., Paquot M., Marlier M. (2001). Les esters de sucres : voies de synthèse et potentialités d’utilisation. Biotechnol. Agron. Soc. Environ. 5, p. 209–219.
Pleiss J., Fischer M., Schmid RD. (1998). Anatomy of lipases binding sites: the scissile fatty acid binding site. Chem. Phys. Lipids 93, p. 67–80.
Ramos ML., Caldeira MM.,Gil VMS (1996). NMR study of uronic acids and their complexation with molybdenum (VI) and tungsten (VI) oxions. Carbohydrate Res. 286, p. 1–15.
Sarney DB., Vulfson EN. (1995). Applications of enzyme to the synthesis of surfactants. Trends Biotechnol. 13, p. 164–172.
Schmidt-Dannert C. (1999). Recombinant microbial lipases for biotechnological applications. Bioorg. Med. Chem. 7, p. 2123–2130.
Seino H., Uchibori T., Nishitani T., Inamasu S. (1984). Enzymatic synthesis of carbohydrate esters of fatty acid (I). Esterification of sucrose, glucose, fructose and sorbitol. J. Am. Oil Chem. Soc. 61, p. 1761–1765.
Tsitsimpikou C., Daflos H., Kolisis FN. (1997). Comparative studies on the sugar esters synthesis catalysed by Candida antarctica and Candida rugosa lipases in hexane. J. Mol. Catal. B: Enzymatic 3, p. 189–192.
Vulfson EN. (1998). Enzymatic synthesis of surfactants, Marcel Dekker, New York, 279 p.
Wessel HP., Bartsch S. (1995). Conformational flexibility in highly sulfated β-D-glucopyranoside derivatives. Carbohydrate Res. 274, p. 1–9.