
Analyse moléculaire de la diversité génétique des dromadaires (Camelus dromedarius) en Tunisie
Institut national agronomique de Tunisie. Laboratoire des Ressources animales et alimentaires. 43, Avenue Charles Nicolle. TU-1082 Tunis (Tunisie). E-mail : ouldahmedmohamed@yahoo.fr
Office de l'Élevage et des Pâturages. 30, Rue Alain Savary. TU-1002 Tunis le Belvédère (Tunisie).
Institut national de Recherche agronomique de Tunisie. Rue Hédi Karray. TU-2049 Ariana (Tunisie).
Banque nationale des Gènes. Boulevard du Leader Arafat. ZI Charguia. TU-1080 Tunis (Tunisie).
Reçu le 31 mars 2009, accepté le 4 février 2010
Résumé
Cette étude s’est intéressée à l’investigation de l’analyse de la diversité et des relations phylogéniques entre les populations de dromadaires dans trois régions arides du Sud tunisien (Kebili, Médenine et Tataouine). Huit marqueurs microsatellites ont été utilisés pour génotyper 90 individus. Au total, 34 allèles ont été détectés dans les trois populations. Le nombre d’allèles par locus varie de 2 à 7, avec une moyenne de 4,25 allèles par locus. Pour chaque population, la moyenne d’allèles par locus est de 3,33 ; 3,71 et 3,87 pour Kebeli, Médenine et Tataouine, respectivement. Le taux de l’hétérozygotie observée est de 0,43 ; 0,50 ; 0,57 et 0,52 pour Kebeli, Médenine, Tataouine et la population totale, respectivement. Ces valeurs sont inférieures à celles de l’hétérozygotie attendue : 0,50 ; 0,57 ; 0,62 et 0,61, respectivement. Les coefficients de consanguinité sont de 15,3 % pour Kebeli, 11,4 % pour Médenine et 8,3 % pour Tataouine. Les F-sat moyens sont FIT = 0,15, FIS = 0,071 et FST = 0,083, ces valeurs sont significativement différentes de zéro (p < 0,05) et suggèrent une différentiation modérée. Un taux de consanguinité de 15 % dans la population totale a été trouvé. Les distances génétiques estimées varient de 0 à 0,9 entre les individus de la population totale. Les distances génétiques entre les paires des populations indiquent 0,104 entre Médenine et Tataouine, 0,280 entre Kebili et Médenine et 0,290 entre Kebeli et Tataouine. Cette matrice de distance permet de distinguer deux entités génétiques : Nefzawa (Kebili) qui inclut les écotypes Merzougui, G’oudi et Mehari et le groupe de Aaradh (Médenine et Tataouine) qui inclut Magherbi et Khaouar. Les résultats de ce travail ne confirment pas la classification actuelle établie par les éleveurs qui divisent la population en cinq écotypes selon des critères sociogéographiques. Ces résultats préliminaires montrent que les microsatellites sont un outil prometteur pour la caractérisation des races animales. Ils indiquent que la population étudiée a une diversité génétique satisfaisante qui peut être utilisée comme base pour aménager des programmes de conservation et de développement durable de l’espèce.
Abstract
Analysis of molecular genetic diversity of dromedaries (Camelus dromedarius) in Tunisia. The objective of this study was to investigate the genetic diversity and relationships among Tunisian camel populations in three different geographical locations (Kebili, Medenine and Tataouine) from Southern arid and semi-arid regions in Tunisia. Eight selected microsatellite markers were used for a sample of 90 dromedary genotypes. A total of 34 alleles were detected in the three populations. The number of alleles per locus varied from two to seven with an average of 4.25 alleles per locus. For each population the average of alleles per locus is 3.33, 3.71 and 3.87 for Kebili, Medenine and Tataouine, respectively. The mean of the observed heterozygosity (Ho) were 0.43, 0.50, 0.57 and 0.52 for Kebili, Medenine, Tataouine and total populations, respectively. These values were lower than expected with heterozygosity (He) values 0.50, 0.57, 0.62 and 0.61, respectively. The average inbreeding coefficient was 15.3% in Kebili, 11.4% in Medenine and 8.3% in Tataouine. The mean estimates of F-statistics were FIT = 0.15, FIS = 0.071 and FST = 0.083. These values were significantly different from zero (p < 0.05) and suggest a moderate differentiation. An inbreeding rate of 15% was found. Estimated genetic distances revealed by the loci varied from 0 to 0.9 between dromedary individuals. The estimated genetic distances pair-wise showed 0.104 among Medenine-Tataouine, 0.280 between Kebili-Medenine and 0.290 between Kebili-Tataouine. The distance matrix was able to distinguish between two separate genetic entities: Nefzawa (Kebili) including Merzougui, G’oudi and M’hari ecotypes and the Aaradh group (Medenine and Tataouine) that includes Maghribi and Khaouar ecotypes. The results of this study did not confirm the present classification established by dromedary herders who divide the population into five different ecotypes, apparently based on the sociogeographical criteria. These preliminary results showed that microsatellites are promising tools for breed characterization. They indicated that the populations under investigation have a high genetic variability and would be suitable as genetic stocks for conservation and sustainable utilization programs.
1. Introduction
1La disponibilité d’informations sur l’histoire des animaux d’élevage et les données sur les rapports génétiques entre les races fournissent un support important au processus de prise de décision pour la conservation des ressources génétiques dans une région donnée. Dans le passé proche, les inférences sur l’histoire des races ont été essentiellement fondées sur des considérations ethnographiques, anthropologiques et archéologiques. Actuellement, ces considérations sont de plus en plus remplacées par les techniques modernes de la génétique moléculaire (Rege et al., 2003). À l’aide de ces techniques moléculaires, le polymorphisme de l’ADN est devenu plus accessible et les méthodes de mesure de ce polymorphisme se sont multipliées : variabilité génétique, consanguinité et flux génétiques (Buchanan et al., 1994 ; Moazami-Goudarzy et al., 1997), identification de gènes d’intérêt économique, empreintes génétiques et cartographie (Bishop et al., 1994). Les marqueurs phénotypiques, biochimiques et, plus récemment, les marqueurs moléculaires au niveau de l’ADN constituent les principales sources de données pour caractériser la diversité génétique et les filiations entre les races. Récemment, les marqueurs moléculaires ont constitué de nouveaux outils pour analyser la diversité génétique. Parmi ces marqueurs, on trouve les microsatellites qui ont vite acquis le statut de marqueurs privilégiés en génétique des populations en raison des avantages qu’ils offrent, notamment en matière de conservation (Canon et al., 2001). La séquence nucléotidique que constitue un microsatellite est composée de répétitions en tandem de trimères, dimères ou même monomères. Très nombreuses, bien réparties sur le génome, ces séquences se caractérisent par un polymorphisme important dû à la variation du nombre de répétitions selon les allèles (Boichard et al., 1998). Vu leur informativité élevée et leur quasi-uniforme distribution dans le génome, les microsatellites représentent les marqueurs idéaux pour la recherche de gènes et l’étude de la diversité génétique. L’utilité des microsatellites pour l’évaluation de la diversité génétique et les études de conservation des races des animaux d’élevage a été confirmée par de nombreux travaux de recherche (Boichard et al., 1998 ; Mburu et al., 2003). Toutefois, les microsatellites sont des marqueurs neutres qui ne correspondent à aucune fonction connue, donc probablement à aucun caractère sélectionné. Certains auteurs (Buchanan et al., 1994 ; MacHugh et al., 1998 ; Saitbekova et al., 1999 ; Hanotte et al., 2000 ; Hanslik et al., 2000 ; Mburu et al., 2003) suggèrent l’emploi de ce type de marqueurs comme l’outil le plus approprié pour différencier les races animales et mettre en évidence la diversité génétique.
2L’objectif de ce travail était d’étudier la diversité génétique au sein de la population de dromadaires de trois régions arides du Sud tunisien (Kebili, Médenine et Tataouine) à l’aide de marqueurs microsatellites. Les principaux écotypes identifiés sont Merzougui, G'oudi et Mhari dans la région Kebili (Nefzawa) et Maghribi et Khaouar dans les régions de Médenine et Tataouine (Aaradh) (Ould Ahmed et al., 2009).
2. Matériel et méthodes
2.1. Prélèvement du sang
3Le sang a été prélevé sur des animaux adultes au niveau de la veine jugulaire. Les prises sanguines ont été effectuées sur l’animal baraqué cou tendu tiré vers l’avant pour faciliter une stase veineuse. L’emploi des tubes stériles sous vide avec bouchons en caoutchouc permet l’utilisation des aiguilles stériles plus fines et moins traumatisantes pour l’animal. Le sang est collecté dans des tubes contenant l’acide éthylène-diamine-tétra-acétique (EDTA), produit permettant la conservation des acides nucléiques du sang pour une longue durée. Pour la collecte proprement dite, l’aiguille est insérée dans la veine jugulaire de l’animal, une fois l’aiguille en place l’écoulement du sang commence puis l’aiguille est introduite dans le tube pour le remplir du sang.
2.2. Extraction de l’ADN génomique
4Le matériel biologique de base pour ce travail est constitué du sang collecté dans des tubes EDTA, puis congelé à -20 °C. Les leucocytes (globules blancs) sanguins représentent la source majeure des acides nucléiques dans le sang. L’extraction de l’ADN génomique a été faite à partir de ce sang entier et congelé. Après une décongélation du sang, on a fait éclater les globules rouges du sang par choc osmotique en les mélangeant à une solution hypotonique ou solution de lyse des globules rouges (SLR). La récupération des globules blancs est faite par centrifugation. Un mélange de détergent de Sodium Dodecyl Sulfate (SDS) est ajouté pour détruire les membranes et de protéinase K pour digérer les protéines associées à l’ADN. La purification de l’ADN de protéines s’est faite par le chloroforme en ajoutant du NaCl pour augmenter la force ionique, puis on précipite l’ADN par l’alcool absolu au froid. L’ADN s’est précipité sous forme de filaments dissous dans une solution tamponnée de conservation, puis est stocké à 4 °C (Annexe 1 : protocole expérimental détaillé de l’extraction de l’ADN).
2.3. Amplification des microsatellites par PCR
5Les conditions d’amplification sont les suivantes : une première dénaturation à 95 °C pendant 10 min suivie de 35 cycles successifs, chaque cycle comprend une succession de trois phases : une dénaturation à 94 °C pendant 45 s, une phase d’hybridation à la température optimale déterminée entre 50-65 °C pendant 1 min selon l’amorce et une élongation à 72 °C pendant 1 min. Enfin, une dernière étape d’élongation à 72 °C pendant 15 min est programmée et une phase de refroidissement à 4 °C. Ces conditions des cycles sont celles utilisées par Mburu et al. (2003). Ainsi, l’optimisation a été faite en fonction d’autres paramètres, à savoir la concentration et le volume. Le volume réactionnel utilisé pour chaque échantillon de 25 µl contient 2,5 mM MgCl2, 0,2 mM dNTP, 0,5 µM de chaque amorce, 1U Taq50 ng.µl-1 ADN et 5 µl Tampon 1X.
2.4. Loci microsatellites amplifiés
6Dans le cadre de ce travail, huit loci microsatellites ont été considérés pour caractériser la variabilité génétique des dromadaires en Tunisie. Ces marqueurs ont été choisis tout en considérant des microsatellites retenus dans d’autres projets de recherche, ainsi que certains recommandés par la FAO (2004) pour analyser la diversité génétique des camélidés. Les marqueurs microsatellites utilisés sont : CVRL01, CVRL02, CVRL05, CVRL06 et CVRL07 (Sasse et al., 2000 ; Mariasegaram et al., 2002), VOLP03 et VOLP32 (Obreque et al., 1998) et YWLL02 (Lang et al., 1996). Ces microsatellites ont été amplifiés par PCR, puis ont été mis à migrer selon une électrophorèse en gel dénaturant de polyacrylamide (PAGE) à 6 % qui permet de distinguer les allèles en fonction de leur taille en paires de bases.
2.5. Électrophorèse sur gel de polyacrylamide
7Morgante et al. (1993) signalent que cette technique permet de séparer des fragments d’ADN même avec une seule base nucléotidique de différence, tel le cas des marqueurs microsatellites. La solution de polyacrylamide à 6 % de volume de 120 ml a été préparée à partir d’une solution à 40 % d’acrylamide, qui est l’unité de base et de bisacrylamide (19:1), d'urée et de TBE. Ces trois solutions sont mélangées et chauffées jusqu’à la dissolution complète de l’urée. La réaction de polymérisation proprement dite s’est faite grâce à l’ajout de deux substances réactives : le TEMED et le persulfate d’ammonium. La migration a été faite pendant 2 h sous 75 W. La coloration s’est faite selon les étapes signalées par Benbouza et al. (2006).
2.6. Méthodes statistiques et analyses moléculaires
8La diversité génétique au sein de la population des dromadaires a été analysée à deux niveaux : la variabilité intrapopulation et la variabilité interpopulation. Le but de cette approche était de caractériser à l’aide d’un ensemble de paramètres la variabilité de la population de dromadaires et la proximité génétique des populations.
9Dans ce travail, on a utilisé la loi de Hardy-Weinberg pour tester si une population est en équilibre. Cette méthode est basée sur la disparité des effectifs de génotypes observés aux effectifs de génotypes théoriques. Le calcul a été effectué à l’aide du logiciel PopGene version 1.31 (Yeh, 1999).
10La phylogénie a été reconstruite en utilisant l’approche Neighbour Joining (N-J) à l’aide du logiciel Darwin version 5.0.155 (Perrier et al., 2006).
11Diversité génétique intrapopulation. Pour décrire la diversité génétique intrapopulation, nous avons calculé six paramètres. Ils sont estimés pour chaque locus et la moyenne est prise sur tous les loci, à l’aide du logiciel PopGene version 1.31 (Yeh, 1999). En termes de variabilité génétique intrapopulation, la comparaison directe des fréquences alléliques n’est pas facile à réaliser. Toutefois, il existe des paramètres susceptibles de synthétiser, moyennant une valeur globale, les informations les plus importantes comme le nombre moyen d’allèles par locus (A), le taux de polymorphisme (P), le taux moyen d’hétérozygotie observé (HO), le taux moyen d’hétérozygotie attendu (HE), le taux moyen d’hétérozygotie attendu non biaisé (Hnb) et l’indice de fixation FIS.
12Nombre moyen d’allèles par locus (A). Ce paramètre traduit la richesse en allèles d’une population, il est calculé selon la formule :

13où a représente le nombre d’allèles à un locus et l le nombre de loci étudiés.
14Taux de polymorphisme (P). C’est le pourcentage de loci polymorphes dans l’échantillon étudié. La probabilité d’observer au moins deux allèles à même locus dépend des fréquences respectives des allèles et aussi de la taille de l’échantillon. Dans le présent travail, un locus est considéré polymorphe dans le cas où l’allèle le plus fréquent a une fréquence inférieure ou égale à 0,95.
15Taux d’hétérozygote observé (HO). C’est la proportion d’individus hétérozygotes au locus K comme dans la formule :
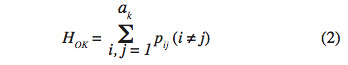
16où Pij est l’estimation de la fréquence du génotype ij au locus k et ak le nombre d’allèles au locus k. Si on considère l locus, le taux d’hétérozygote observé (HO) est la moyenne de (HOK) suivant l’équation :
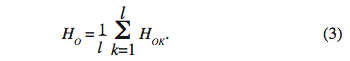
17Taux d’hétérozygote attendu (HE). Un taux d’hétérozygotie attendu (HE) peut être calculé, sous l’hypothèse d’équilibre de Hardy-Weinberg, à partir des fréquences alléliques déterminées pour chaque locus à l’aide de la formule :

18où pi est la fréquence du ième allèle à ce locus.
19Le taux moyen d’hétérozygotie est l’indice le plus satisfaisant de la diversité génétique. Sa valeur numérique dépend du nombre de loci polymorphes et de la structure génotypique de chacun d’eux. Nei (1978) propose d’utiliser un estimateur non biaisé (Hnb) ou la diversité génique lorsque le nombre d’animaux testés est faible. Celle-ci est définie comme étant la probabilité de tirer, au hasard, deux allèles différents à un même locus.
20L’estimation non biaisée est calculée selon la formule :
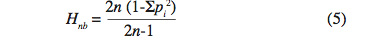
21où n est le nombre d’individus étudiés.
22Les paramètres ainsi définis peuvent décrire, jusqu’à un certain niveau, la diversité génétique des populations ainsi que celle des individus qui les composent. Certes, la variabilité génétique d’un individu demeure inchangée tout au long de sa vie, à l’exception d’éventuelles mutations. Toutefois, la constitution génétique d’une population présente une possibilité de variation dans le temps. En effet, les populations subissent des forces qui déterminent leur constitution génétique et tendent à la maintenir ou à la modifier. La conséquence de ces forces évolutives est de faire varier le taux d’hétérozygotie de la population par rapport à l’équilibre de Hardy-Weinberg. Il devient alors intéressant de pouvoir quantifier ces écarts entre l’hétérozygotie observée et celle attendue à l’équilibre. De plus, deux populations d’une même espèce peuvent être soumises à des facteurs évolutifs différents et voir leur constitution génétique se différencier l’une de l’autre, ce qui peut produire un effet à la fois sur les fréquences alléliques et sur la relation entre l’hétérozygotie observée et celle attendue. Les écarts par rapport à l’équilibre de Hardy-Weinberg sont quantifiés à l’aide de la statistique hiérarchique des coefficients F.
23Indice de Fixation FIS. Ce paramètre mesure l’écart entre la population d’individus trouvés à l’écart hétérozygote HO et le taux d’hétérozygote attendu, il est appelé aussi l’écart à la panmixie et se calcule comme il est mentionné dans la formule :

24Au locus donné, dans une population d’individus diploïdes, l’association de deux gamètes pour former les individus se fait au hasard par rapport aux génotypes de ces gamètes, la structure génétique de cette population est appelée structure de Hardy-Weinberg. L’indice de fixation est l’écart à la structure de Hardy-Weinberg, il varie de -1 à +1 et permet de connaitre le déficit en hétérozygotes par population, par locus et pour l’ensemble des loci. F est positif quand la population présente un déficit en hétérozygotes par rapport à l’équilibre panmictique et négatif dans le cas contraire. Un certain nombre de facteurs contribuent à cet écart : consanguinité, dérive, sélection, différenciation, etc.
25Diversité génétique interpopulation. Pour décrire la diversité génétique entre les populations, nous avons utilisé dans la présente étude trois paramètres.
26Distance génétique. La distance génétique entre deux échantillons est définie comme la proportion d’éléments génétiques qu’ils n’ont pas en commun. D = 1 si et seulement si les deux échantillons n’ont pas d’éléments génétiques en commun.
27Dans le cadre de ce travail, les distances génétiques entre paires de populations ont été calculées selon l’approche classique basée sur les fréquences alléliques dans chaque population en retenant la distance de Nei (1978). Cette distance présente des propriétés spécifiques et reste appropriée pour un modèle particulier d’évolution. Elle considère un modèle mutation-dérive et elle est destinée à mesurer le nombre moyen de différences de codons entre populations après leur divergence. Cette distance est la plus utilisée dans les recherches sur la diversité génétique.
28F statistiques de Wright. Dans une population subdivisée, il existe trois niveaux de complexité : les individus (I), les sous-populations (S) et la population totale (T). Dans ce travail, les populations relatives aux régions représentent les sous-populations et l’ensemble des populations représente la population globale. Pour mesurer l’organisation de la diversité génétique dans une population, Wright (1978) a défini l’hétérozygotie de chacun de ces trois niveaux respectivement par les paramètres suivants : HI, HS et HT. Le premier paramètre HI correspond à l’hétérozygotie moyenne des individus sur l’ensemble des sous-populations. Il représente également l’hétérozygotie moyenne observée pour l’ensemble des gènes (ou loci) d’un individu. C’est aussi la probabilité d’hétérozygotie en un locus pris au hasard. Ainsi, si Hi est l’hétérozygotie observée dans la ième sous-population, on aura, pour X sous-populations, la formule :

29Le second paramètre HS indique l’hétérozygotie attendue par individu pour chaque sous-population en la supposant à l’équilibre Hardy Weinberg. Il représente aussi l’hétérozygotie attendue dans une sous-population supposée à l’équilibre Hardy-Weinberg où pi est la fréquence du ième allèle. Soit pour la Sème :
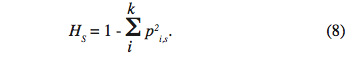
30On notera HS* la moyenne des HS sur les X sous-populations :
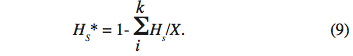
31Enfin, le dernier paramètre HT représente l’hétérozygotie attendue par individu, en supposant la population globale à l’équilibre Hardy-Weinberg. En d’autres termes, c’est l’hétérozygotie attendue si toutes les sous-populations étaient regroupées en une seule unité panmictique. Si l’on note pi* la fréquence moyenne de l’allèle Ai sur l’ensemble des X sous-populations, on obtient :
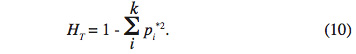
32Trois indices sont générés à partir de ces hétérozygoties : FIS, FST et FIT. Ces derniers mesurent l’écart de l’hétérozygotie par rapport à l’équilibre Hardy-Weinberg (EHW) à différents niveaux. Le premier indice FIS est défini par la relation :
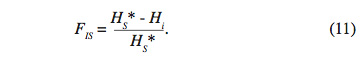
33Cet indice, appelé coefficient de consanguinité, mesure la réduction éventuelle de l’hétérozygotie des individus à l’intérieur de leur sous-population. En cas de consanguinité, cet indice est positif et indique un déficit en hétérozygotie. Évidemment, il prend la valeur zéro si les sous-populations sont à l’EHW. En revanche, s’il est négatif, les populations présentent un excès d’hétérozygotie.
34Entre les sous-populations et la population totale, l’effet de la subdivision est exprimé par un indice similaire :

35Ce paramètre, appelé indice de fixation, correspond à la réduction de l’hétérozygotie dans les sous-populations liée aux différences de fréquences alléliques moyennes. Cet indice renseigne sur la différenciation et l’effet de subdivision des populations. Il prend la valeur zéro lorsque toutes les sous-populations ont les mêmes fréquences alléliques et sont à l’EHW. Dans le cas contraire, l’effet Wahlund implique que HT soit plus grand que HS* et donc FST sera positif. Enfin, la réduction de l’hétérozygotie entre l’individu et la population globale théorique est donnée par la formule :

36Ces trois indices sont liés par la relation :
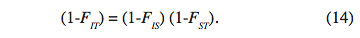
37Si toutes les sous-populations sont bien à l’EHW, on aura FIS = 0 et par conséquent FST = FIT. Par ailleurs, si elles sont toutes à l’EHW et ont les mêmes fréquences alléliques, alors les trois indices seraient nuls. Dans ce cas, la division en sous-populations n’existe plus et la population globale est à l’EHW. Comme il a été souligné, l’indice de fixation FST permet de quantifier le degré de diversification génétique entre les populations. Les paramètres FIT, FIS et FST désignent respectivement les indices de fixation d’un individu de la population, d'un individu d’une sous-population et d'une sous-population. FIT et FIS mesurent la corrélation entre les gamètes d’un même individu tiré au hasard respectivement dans une sous-population et dans la population totale. FIS permet de mesurer le déficit local moyen d’hétérozygotes par rapport à la structure de Hardy-Weinberg. FIT permet de mesurer le déficit global d’hétérozygotes dans l’ensemble de la population. Alors que FST représente la corrélation entre deux gamètes tirés au hasard dans deux sous-populations différentes et renseigne sur le niveau de différenciation ou l’individualisation des sous-populations, déficit connu sous le nom de « effet de Wahlund », 0 ≤ FST ≤ 1 (Nei, 1973). Le logiciel Fstat version 2.9.3.2 (Goudet, 2001) a été utilisé pour le calcul de ces indices ; le niveau du test de signification est : p < 0,05.
38Flux des gènes. La différenciation génétique entre populations est favorisée par la dérive et limitée par les flux génétiques entre les populations. Le nombre de migrants effectifs par génération (Nm) est relié à la différenciation génétique FST par la relation :
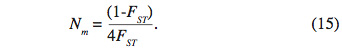
39Ce paramètre a été calculé à l’aide du logiciel Genetix version 4.05.2 (Belkhir et al., 2004).
3. Résultats et discussion
3.1. Diversité génétique intrapopulation
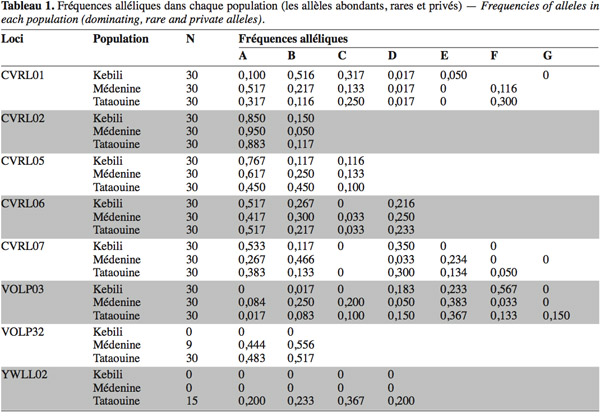
40Fréquences alléliques. D’une manière générale, la distribution des allèles, dont le nombre varie de 2 pour les loci CVRL02 et VOLP32 à 7 pour le locus VOLP03, est assez identique dans la plupart des cas, les allèles les plus fréquents étant toujours les mêmes. C’est notamment le cas de l’allèle A (Tableau 1) qui apparait souvent, sinon toujours, le plus abondant dans les différents loci de toutes les populations étudiées. Les fréquences alléliques calculées pour chaque locus et chaque population varient de 0,017 pour plusieurs allèles dans quelques populations à 0,950 pour l’allèle A du CVRL02 au niveau de la population de Médenine. Le tableau 1 présente les fréquences des allèles des populations. Au niveau du locus CVRL02 qui compte deux allèles, la dominance de l’allèle A est assez remarquable et sa fréquence est supérieure à 0,850 dans toutes les populations. Quant à VOLP32, il exhibe deux allèles, qui ne sont présents que dans les populations de Médenine et de Tataouine. Ces dernières régions sont géographiquement très proches l’une de l’autre. Le schéma de distribution de ces deux allèles présente une similarité dans les deux populations. La présence exclusive du locus YWLL02 avec ses 4 allèles chez la population de Tataouine a été notée, avec une légère différence des fréquences de ces 4 allèles en faveur de l’allèle C.
41Richesse allélique. La richesse allélique d’une population, définie comme le nombre d’allèles présents à un locus donné, est connue pour dépendre de la taille de l’échantillon, puisque les chances de découvrir un nouvel allèle augmentent chaque fois qu’un nouvel individu est observé (Foulley et al., 2006). Dans ce travail, le nombre moyen d’allèles par locus varie de 3,33 pour Kebili à 3,87 pour Tataouine. Il est de 4,25 pour la population globale, ce qui est relativement important.
42Le taux de loci polymorphes. Tous les loci sont révélés polymorphes à 100 % au seuil de 95 % dans les trois populations. Ce résultat indique l’efficacité des loci microsatellites utilisés pour l’analyse de la diversité génétique des populations étudiées.
43Hétérozygotie. Les taux d’hétérozygotie observée et attendue ont été calculés pour chaque locus et population sous l’hypothèse d’équilibre de Hardy-Weinberg. Afin d’estimer l’importance du polymorphisme génétique, nous avons comparé les deux taux d’hétérozygotie. Pour la population totale sur la base de multilocus, l’hétérozygotie attendue non biaisée (0,61) est supérieure à celle observée (0,52), traduisant un écart positif suggérant un déficit en hétérozygotie dans la population (Tableau 2). La valeur de (Hnb = 0,61) est comparable à celle des dromadaires en Afrique du Sud (0,604), inférieure à la valeur enregistrée chez la population soudanaise (0,68) (Nolte et al., 2005) et elle est strictement supérieure aux valeurs trouvées chez les populations du Kenya (0,53) et des Émirats arabes (0,51) (Mburu et al., 2003). Cependant, HO = 0,52 a été moins importante que d’autres valeurs trouvées dans d’autres études. Vijh et al. (2007) signalent que les valeurs de HO pour les races camelines indiennes sont de 0,58, 0,57, 0,56, et 0,60 pour Bikaneri, Jaisalmeri, Kutchi et Mewari, respectivement.
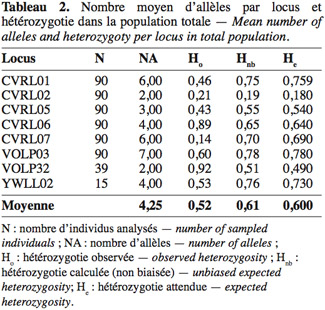
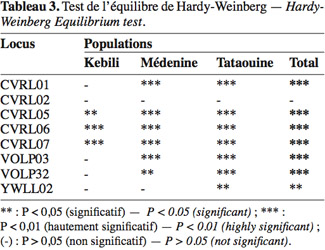
44Remarquons que certains loci sont en écart significatif par rapport à l’équilibre de Hardy-Weinberg ; le tableau 3 montre les résultats du test d’équilibre vérifié par le logiciel Popgene. Notons que tous les loci sont en déséquilibre dans la population totale, à l’exception du locus CVRL02 qui vérifie aussi bien l’équilibre dans toutes les populations individuelles.
45Indice de fixation FIS. Cet indice est la mesure de l’écart entre la population d’individus trouvés à l’état hétérozygote (HO) et l’hétérozygote attendu (HE). Les trois populations présentent des indices de fixation (FIS) positifs plus ou moins élevés compris entre (FIS = 0,083) pour Tataouine et (FIS = 0,153) pour Kebili, alors que Médenine (FIS = 0,114) prend une valeur intermédiaire entre les deux. Ces indices positifs traduisent aussi un déficit en hétérozygotes dont les loci CVRL07 et CVRL01 sont en partie responsables, car lesdits loci présentent des FIS souvent supérieurs à 0,35 dans toutes les populations analysées. Les trois populations présentent des coefficients de consanguinité dont les valeurs sont 15,3 %, 11,4 % et 8,3 % pour Kebili, Médenine et Tataouine, respectivement. Mis à part le hasard, trois principaux facteurs pourraient expliquer ce déséquilibre observé. Il s’agit de facteurs génétiques, l’existence d’allèles nuls et l’effet de Wahlund (Jordana et al., 2003). En ce qui concerne les causes génétiques, il est bien connu que la consanguinité (accouplement entre un individu et ses ascendants, ses collatéraux et/ou ses descendants) modifie les fréquences génotypiques. La conséquence en est une perte de variabilité génétique au cours des générations. Le second facteur pourrait être inhérent à l’existence d’allèles nuls, allèles ne donnant lieu par PCR à aucune amplification. Une délétion au niveau des amorces ou une mutation dans les séquences flanquantes du microsatellite pourraient entrainer la présence d’allèles nuls. Enfin, le dernier facteur fait référence à la présence de sous-populations à l’intérieur de chaque population (région) pouvant induire l’effet de Wahlund. À l’échelle de la population totale, les loci affichent un excès d’homozygotes assez important avec FIS = 0,33 pour CVRL01 et FIS = 0,78 pour CVRL07. L’excès en homozygotie ou hétérozygotie observé par rapport à l’homozygotie ou l’hétérozygotie attendues sous l’hypothèse de Hardy-Weinberg a été testé pour chaque locus et population. Ainsi, il apparait que certaines populations vis-à-vis de certains loci présentent des excès d’homozygotes significatifs par rapport aux proportions de l’équilibre de Hardy-Weinberg. D’autres populations ont des excès en hétérozygotes significativement différents des proportions de l’équilibre de Hardy-Weinberg et bien sûr, quelques populations ont montré l’équilibre panmictique au niveau de certains loci. Les valeurs moyennes du FIS : 0,153, 0,114 et 0,083 pour Kebili, Médenine et Tataouine, respectivement, sont toutes positives, suggérant ainsi un déficit en hétérozygotes chez toutes les populations camelines étudiées. L’indice moyen pour la population globale est de 0,071, indiquant un déficit d’hétérozygotes relativement modéré.
3.2. Diversité génétique interpopulation
46Indice de fixation ou F statistiques. Les indices de fixation ont été calculés pour tous les loci. Les valeurs de FIS varient de -0,84 pour VOLP32 à 0,78 pour CVRL07 et celles du FIT varient de 0,364 au locus CVRL06 à 0,816 pour YWLL02. La valeur moyenne de FIS = 0,071 indique un déficit d’hétérozygotes moins important au niveau des populations prises isolément que dans la population globale (FIT = 0,150), probablement dû à l’effet Wahlund. La valeur de FIT indique un déficit global d’hétérozygotes de 15 %, en tenant compte des trois populations étudiées. Quant à la différenciation génétique FST, elle est relativement élevée pour YWLL02 et VOLP32. En revanche, FST affiche des valeurs faibles aux loci CVRL02 et CVRL06. La différenciation moyenne entre les populations est de FST = 0,083, ce qui peut être considéré comme une valeur globalement modérée, indiquant l’origine de la variation génétique totale dans l’espèce. Rappelons que la diversité génétique totale est la somme de la diversité génétique intrapopulation et de la diversité génétique interpopulation. La valeur FST = 0,083 détermine qu’une grande part (91,7 %) de la variabilité génétique totale est expliquée par la variation intrapopulation et que 8,3 % de cette variabilité est attribuée aux différences entre populations de l’espèce. Ce niveau de différenciation génétique reste, globalement, semblable aux niveaux cités dans d’autres études pour d’autres espèces domestiques : 8,2 % pour les races camelines indiennes (Vijh et al., 2007), 8 % pour les équidés en Espagne (Canon et al., 2000) et 10 % entre les populations bovines européennes (MacHugh et al., 1998). FST a été calculé entre les populations ; le paramètre de différenciation génétique FST = 0,031 entre Médenine et Tataouine traduit l’absence de structuration géographique et génétique entre ces deux populations qui apparaissent homogènes. Ce net rapprochement suppose des échanges d’animaux entre ces deux régions. En revanche, on retrouve une distinction entre la population de Kebili et celle des deux autres régions (FST = 0,131 entre Médenine et Kebili et FST = 0,108 entre Kebili et Tataouine), qui peut s’expliquer par l’isolement de cette région.
47Distance génétique et construction des dendrogrammes. La matrice des distances estimées entre les individus de la population totale a servi pour la construction des dendrogrammes. Ces distances varient de 0 à 0,9, ce qui montre une large variabilité génétique au sein de la population cameline étudiée. La distance nulle entre deux individus suggère une similarité vis-à-vis des loci étudiés. Par contre, la distance la plus élevée traduit la divergence entre ces individus.
48L’examen du dendrogramme de la population totale (Figure 1) permet de distinguer trois groupes principaux qui, à leur tour, présentent des sous-groupes. L’analyse de l’arbre montre que le regroupement des individus se fait indépendamment de l’origine géographique et du nom ethnique de l’écotype. Cette répartition sur l’arbre phylogénique peut être expliquée par l’existence d’une large base génétique commune entre les différentes populations et écotypes et ce, en dépit de la divergence géographique et phénotypique, en plus de l’absence d’un programme ciblé d’amélioration génétique.
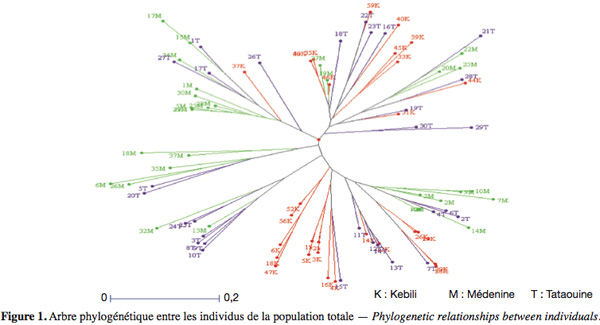
49Pour étudier la structuration de la diversité génétique au sein des populations individuelles, la distance de Nei (1978) a été estimée entre les paires de populations. La matrice des distances génétiques entre les populations indique une variation de 0,104 entre Médenine et Tataouine ; 0,29 entre Kebili et Tataouine ; 0,28 entre Kebili et Médenine. Ces valeurs relativement peu élevées indiquent que les populations présentent une ressemblance génétique et appartiennent à un même groupe génétique.
50En examinant le dendrogramme des populations, on peut remarquer qu’en général les regroupements des populations sont en relation avec des proximités géographiques. Nous constatons que le rameau de Nefzawa (Kebili) constitue un groupe isolé. Cependant, le rameau de l’Aaradh (Médenine et Tataouine) forme un groupe divisé en deux classes (Figure 2).
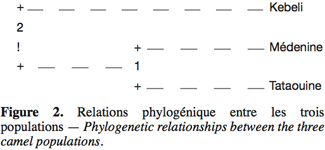
51L’analyse des populations prises individuellement montre bien que les subdivisions de chaque population ne correspondent pas aux écotypes et que le regroupement se fait indépendamment de ces écotypes. Ce qui mène à l’hypothèse que les écotypes identifiés et nommés par les éleveurs ne constituent pas des entités génétiques bien individualisées. Cette hypothèse peut être consolidée par les résultats obtenus dans le chapitre précédent comme les pratiques des croisements incontrôlés, des choix et d’utilisation des mâles reproducteurs (85 % des géniteurs sont choisis du troupeau lui-même et utilisés pendant une durée moyenne de 7 ans) et la consanguinité, tous ces facteurs limitent significativement la différenciation, outre la faible taille échantillonnée de certains écotypes étudiés.
52Flux des gènes. Les flux des gènes (Nm) entre les paires des populations représentent une valeur relativement importante (7,73) entre Médenine et Tataouine, alors que moins importante chez les autres populations (1,65 entre Kebili et Médenine et 2,06 entre Kebili et Tataouine). Globalement, les valeurs traduisent un échange de gènes assez important entre ces trois populations. Ces résultats de Nm confirment ceux de distances et différenciations génétiques.
4. Conclusion
53Ces résultats présentés ne constituent qu’une modeste contribution à la caractérisation des dromadaires en Tunisie. Ils représentent également autant d’acquis pour la mise en œuvre d’un programme de développement de cette espèce dans le pays. En outre, ce travail pourrait ainsi servir de base à des programmes plus conséquents d’études de la diversité génétique des différentes populations de dromadaires en Tunisie. D’une manière générale, la présente étude aura permis de mettre en évidence chez les dromadaires en Tunisie un niveau élevé de diversité génétique, principalement d’origine intrapopulation (92 %). Bien que cette population présente un taux de consanguinité de 15 %, les indices de différenciation montrent que les populations partagent une grande base génétique, malgré l’apparition de différences suivant la région. En effet, la population semble menacée par l’absence de gestion des généalogies dans les troupeaux et par des croisements consanguins qui pourraient entrainer une évolution de sa structure génétique vers l’homozygotie. Plusieurs facteurs dont les pratiques des éleveurs, le statut actuel de l’espèce et son histoire évolutive expliquent sa structuration génétique actuelle. Le maintien de la variation génétique existante au sein de l’espèce constituera une étape importante dans le cadre de la conservation des ressources génétiques animales.
Bibliographie
Belkhir K. et al., 2004. GENETIX 4.05.2. Logiciel sous Windows TM pour la génétique des populations. Montpellier, France : CNRS UMR 5000, Laboratoire Génome, Populations, Interactions, Université de Montpellier II, http://www.univ-montp2.fr/~genetix/genetix/constr.htm#download, (02.05.08).
Benbouza H., Jacquemin J.-M., Baudoin J.-P. & Mergeai G., 2006. Optimization of a reliable, fast, cheap and sensitive silver staining method to detect SSR markers in polyacrylamide gels. Biotechnol. Agron. Soc. Environ., 10(2), 77-81.
Bishop M.D. et al., 1994. A genetic linkage map for cattle. Genetics, 136, 619-639.
Boichard D., Leroy P., Leveziel H. & Elsen J.-M., 1998. Utilisation des marqueurs moléculaires en génétique animale. INRA Prod. Anim., 11(1), 67-80.
Buchanan F. et al., 1994. Determination of evolutionary relationships among sheep breeds using microsatellites. Genomics, 22, 397-403.
Canon J. et al., 2000. The genetic structure of Spanish Celtic horse breeds inferred from microsatellite data. Anim. Genet., 31, 39-48.
Canon J. et al., 2001. Genetic diversity measures of local European beef cattles for conservation purposes. Genet. Sel. Evol., 33(3), 311-332.
Goudet J., 2001. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2), http://www.unil.ch/izea/softwares/fstat.html, (02.05.08).
Foulley J.L. & Ollivier L., 2006. Diversité génétique et richesse allélique : concepts et application à des races bovines. In : Treizièmes rencontres autour des recherches sur les ruminants, 6-7 décembre 2006, Paris, France, 227-230.
Hanotte O. et al., 2000. Geographic distribution and frequency of a taurine Bos taurus and an indicine Bos indicus Y specific allele amongst sub-Saharan African cattle breeds. Mol. Ecol., 9, 387-396.
Hanslik S., Harr B., Brem G. & Schlotterer C., 2000. Microsatellite analysis reveals substantial genetic differentiation between contemporary New World and Old World Holstein Friesian populations. Anim. Genet., 31, 31-38.
Jordana J. et al., 2003. Genetic structure of eighteen local South European beef cattle breeds by comparative F-statistics analysis. J. Anim. Breed. Genet., 120, 73-87.
Lang K.D.M., Wang Y. & Plante Y., 1996. Fifteen polymorphic dinucleotide microsatellites in llamas and alpacas. Anim. Genet., 27, 285-294
MacHugh D.E, Loftus R.T., Cunningham P. & Bradley D.G., 1998. Genetic structure of seven European cattle breeds assessed using 20 microsatellite markers. Anim. Genet., 29, 333-340.
Mariasegaram M. et al., 2002. Isolation and characterisation of eight microsatellite markers in Camelus dromedaries and cross-species amplification in C. bactrianus and Lama pacos. Anim. Genet., 33, 385-387.
Mburu D.N. et al., 2003. Genetic diversity and relationships of indigenous Kenyan camel populations: implications for their classification. Anim. Genet., 34, 26-32.
Moazami-Goudarzy K., Laoe D., Furet J.P. & Grosclaude F., 1997. Analysis of genetic relationships between 10 cattle breeds with 17 microsatellites. Anim. Genet., 28, 338-345.
Morgante M. & Olivieri A.M., 1993. PCR-amplified microsatellites as markers in plant genetics. Plant J., 3(1), 175-182.
Nei M., 1973 Analysis of gene diversity in subdivided populations. Proc. Natl Acad. Sci. USA, 70, 3321-3323
Nei M., 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89, 583-590.
Nolte M., Kotze A., Van der Bank F.H. & Grobler J.P., 2005. Microsatellite markers reveal low genetic differentiation among South American Camelus dromedrius populations. South Afr. J. Anim. Sci., 35, 152-161.
Obreque V. et al., 1998. Characterisation of 10 polymorphic alpaca dinucleotide microsatellites. Anim. Genet., 29, 461-462.
Ould Ahmed M., Ben Salem F., Bedhiaf S.& Djemali M., 2009. Camel genetic resources in Tunisia. In: Proceeding of the 2nd Conference of the International Society of Camelid Research and Development, 12-14 March, Djerba, Tunisia, 2009.
Perrier X. & Jacquemoud-Collet J.P., 2006. DARwin software version 5.0.155, http://darwin.cirad.fr/darwin, (02.05.08).
Rege J.E.O. & Gibson J.P., 2003. Animal genetic resources and economic development: issues in relation to economic valuation. Ecol. Econ., 45, 319-330.
Saitbekova N., Gaillard C., Obexer-Ruff G. & Dolf G., 1999. Genetic diversity in Swiss goat breeds based on microsatellite analysis. Anim. Genet., 30, 36-41.
Sasse J. et al., 2000. South American camelid microsatellite amplification in Camelus dromedaries. Anim. Genet., 31, 75-76.
Vijh R.K., Tantia M.S., Mishra B. & Bharani Kumar S.T., 2007. Genetic diversity and differentiation of dromedarian camel of India. Anim. Biotechnol., 18, 81-90.
Wright S., 1978. Evolution and the genetics of populations.Variability within and among natural populations. Chicago, IL, USA: University of Chicago Press.
Yeh F. et al. POPGENE, version 1.31, 1999. A Microsoft Windows based freeware for population genetic analysis. Edmonton, AB, Canada: University of Alberta.
Om dit artikel te citeren:
