


- Portada
- Volume 21 (2017)
- Numéro 2
- Biochemical, biophysical, and thermal properties of alkaline phosphatase from thermophile Thermus sp. NTU-237


Vista(s): 3908 (14 ULiège)
Descargar(s): 482 (2 ULiège)

Biochemical, biophysical, and thermal properties of alkaline phosphatase from thermophile Thermus sp. NTU-237

Notes de la rédaction
Received on August 23, 2016; accepted on May 2, 2017
This article is distributed under the terms and conditions of the CC-BY License (http://creativecommons.org/licenses/by/4.0)
Résumé
Propriétés biochimiques, biophysiques et thermiques de la phosphatase alcaline de la souche thermophile Thermus sp. NTU-237
Description du sujet. Les phosphatases alcalines (APases) sont fréquemment employées comme marqueurs non radioactifs pour la détection spécifique de protéines ou d’ADN cibles en médecine clinique et en biologie moléculaire. Toutefois, leur application en biotechnologie industrielle nécessite une bonne stabilité tant thermique que de conservation. Nos recherches préliminaires ont montré que l’APase de Thermus sp. NTU-237 (TsAPase) est thermostable et présente une haute activité. Il est donc intéressant d’établir les conditions optimales de son emploi.
Objectifs. Caractériser l’APase de la souche thermophile Thermus sp. NTU-237 et évaluer ses potentialités d’application.
Méthode. Le gène de l’APase de Thermus sp. NTU-237 a été cloné pour être exprimé dans Escherichia coli. Cela a permis l’étude des effets sur l’activité enzymatique TsAPase, de la température, du tampon, des teneurs en glycérol, en dodécyl sulfate de sodium (SDS) et en NaCl pour cerner les conditions optimales d’essai. De plus, les potentialités d’application des TsAPase ont été investiguées au moyen du substrat chromogène 5-bromo-4-chloro-3-indolyl phosphate/nitro bleu de tétrazolium (BCIP/NBT).
Résultats. La TsAPase recombinante fonctionne comme un dimère d’une masse moléculaire de 109 kDa. Les alignements de séquences de la TsAPase avec d’autres APases thermophiles connues et l’APase d’E. coli dénotent une bonne conservation des acides aminés impliqués dans l’activité catalytique et dans des fonctions de liaison à certains résidus. Cette découverte suggère que le mécanisme catalytique des TsAPases est identique à celui d’autres APases. L’activité TsAPase s’est avérée être inhibée par le glycérol et le SDS, alors qu’elle s’est accrue en présence de NaCl et de tampon Tris-HCl. Les paramètres cinétiques Km, kcat, et Vmax ont été établis à 81 µM, 6,08 s-1 et 6,76 U·mg-1, respectivement. La température optimale de la TsAPase était de 80 °C. La TsAPase est stable à température ambiante durant plus de 10 jours. De plus, la TsAPase s’est montrée capable de catalyser la déphosphorylation du BCIP, provoquant par la même occasion le développement à température ambiante de la couleur bleue du substrat transformé.
Conclusions. Les résultats rapportés illustrent les potentialités d’utilisation de la TsAPase, que ce soit dans le domaine médical ou en recherche fondamentale.
Abstract
Description of the subject. Alkaline phosphatases (APases) are commonly used as nonradioactive markers for detecting specific proteins or DNA targets in clinical medicine and molecular biology. However, their applications in the biotechnology industry require thermostability and storage stability. Our preliminary study revealed that APase from Thermus sp. NTU-237 (TsAPase) is thermostable and exhibits high activity. Therefore, it is desirable to establish the optimal conditions for its application.
Objectives. To characterize APase from thermophile Thermus sp. NTU-237 and to evaluate its potential applications.
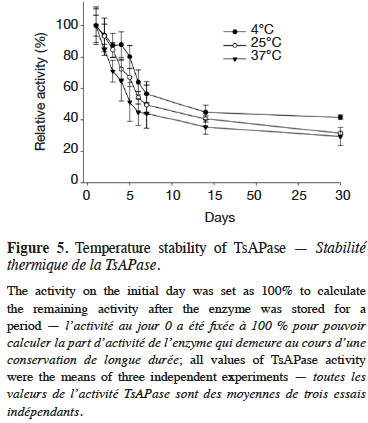
Method. The APase gene of Thermus sp. NTU-237 was cloned and expressed in Escherichia coli. Subsequently, the effects of buffer, glycerol, sodium dodecyl sulfate (SDS), NaCl, and temperature on enzyme activity were studied to establish the optimal conditions for TsAPase assays. In addition, the potential for application of TsAPase was evaluated using the 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) active staining method.
Results. Recombinant TsAPase was identified as having a dimeric structure and a molecular mass of 109 kDa. Sequence alignment analysis of known thermophilic APases with TsAPase and E. coli APase revealed that catalytic and binding residues were highly conserved. This finding suggested that the catalytic mechanism of TsAPase is the same as that of other APases. TsAPase activity was inhibited by glycerol and SDS but enhanced by NaCl and Tris-HCl buffer. The kinetic parameters Km, kcat, and Vmaxwere determined to be 81 µM, 6.08 s-1, and 6.76 U·mg-1, respectively. The optimum temperature of TsAPase was 80 °C, and TsAPase was stable at room temperature for more than 10 days. Moreover, TsAPase could catalyze the dephosphorylation of BCIP and could elicit blue color development by the BCIP/NBT reaction kit at room temperature.
Conclusions. These results illustrate that TsAPase has potential for application in medical or basic research.
Tabla de contenidos
1. Introduction
1Alkaline phosphatase (APase, EC 3.1.3.1) is an orthophosphate monoester phosphohydrolase that catalyzes the hydrolysis of phosphomonoester to produce free inorganic phosphate or to transfer the phosphoryl group to other alcohols (Rodrigues et al., 2006; Barbosa et al., 2008; Deng et al., 2015; Divya et al., 2016). APase is present in all organisms and plays an important role in fundamental biochemical processes, particularly in phosphate transportation and metabolism (Trowsdale et al., 1990; Ásgeirsson & Andrésson, 2001; Deng et al., 2015). Currently, in clinical medicine and molecular biology, APases are extensively applied for detecting specific proteins or DNA through APase conjugation to the targets (Banik & Pandey, 2009; Liang et al., 2013; Claussen et al., 2015). However, such applications require thermostable APase, given that thermostable enzymes are required to maintain almost maximum activity during the incubation and hybridization of APase-conjugated proteins or DNA at high temperatures or when APase-conjugated probes are washed under stringent conditions (with detergents or some denaturing agents). Therefore, the search for thermostable APases has attracted much attention in recent years.
2Since the 1960s, numerous APases have been characterized. Of these APases, Escherichia coli APase is the most extensively studied to date; several studies have investigated its biosynthesis, structure, catalytic properties, and catalytic mechanism (Coleman, 1992; Karamyshev et al., 1998; Claussen et al., 2015; Peck et al., 2016). However, E. coli APase or calf intestine phosphatase possesses low thermostability and a short shelf life, thus restricting their further application (Guimaraes et al., 2001; Li et al., 2007). Consequently, thermostable enzymes have attracted more attention. To date, numerous thermostable APases have been isolated and characterized from various thermophiles, including Thermus species (Kim et al., 1997; Pantazaki et al., 1998; Yuan et al., 1998), Thermotoga neapolitana (Dong & Zeikus, 1997), Meiothermus ruber (Yurchenko et al., 2003), Bacillus stearothermophilus (Mori et al., 1999), Scytalidium thermophilum (Guimaraes et al., 2001), Pyrococcus abyssi (Zappa et al., 2001; Minamihata et al., 2012), and Bacillus licheniformis (Banik & Pandey, 2009; Divya et al., 2016). However, these thermostable APases exhibit drawbacks such as low activity, low thermostability, or low tolerance to alkaline pH and detergents.
3A previous study found that APase from Thermus sp. NTU-237 (TsAPase) is thermostable and exhibits high activity against p-nitrophenyl phosphate (pNPP) at alkaline pH (Chiang, 2010). This enzyme has potential applications at high temperature and alkaline pH. However, no study has reported its biochemical, biophysical, and thermal properties and potential applications for detecting targets. In this study, for the preliminary characterization of thermostable TsAPase, the APase gene of Thermus sp. NTU-237 (TsAPase gene) was cloned using primers designed according to our sequence deposited in Genbank (Accession No. KX267839) and was expressed in E. coli.
2. Materials and methods
2.1. Chemicals and strains
4All chemicals and reagents were purchased from Sigma-Aldrich Fine Chemicals (St. Louis, MO, USA), and culture media were provided by Becton, Dickinson and Company (Franklin Lakes, NJ, USA). Primers were synthesized by Mission Biotechnology (Taipei, Taiwan). All other chemicals used for DNA manipulation and protein analysis were purchased from New England Biolabs (Ipswich, MA, USA), Bio-Rad Laboratories (Hercules, CA, USA), or GE Healthcare (Wauwatosa, WI, USA). Bacterial strains were obtained from the Bioresource Collection and Research Center, FIRDI (Hsinchu, Taiwan).
2.2. DNA manipulation
5In this study, the TsAPase gene was cloned. The genomic DNA of Thermus sp. NTU-237 was prepared using a previously described method (Sambrook et al., 2001). A pair of primers was designed according to the DNA sequence deposited in GenBank (Accession No. KX267839). The primers TsAPase-Nter (5'-GGG GGA TCC AAG CGA AGG GAC ATC CTG-3’) containing a BamHI site and TsAPase-Cter (5'-GGG AAG CTT TTA GGC CCA GAC GTC CTC-3’) containing a HindIII site were used to amplify the TsAPase gene, according to the standard procedure (Sambrook et al., 2001). The amplified gene fragment and the pQE30 vector were digested using restriction enzymes and were ligated using ligase, producing the pQE-TsAPase plasmid. Subsequently, recombinant TsAPase was 6 × His-tagged at the N-terminus. The constructed expression plasmid of pQE-TsAPase was transformed into E. coli for TsAPase overproduction.
2.3. Protein expression, purification, and enzyme assay
6Escherichia coli cells harboring the pQE-TsAPase plasmid were cultured in Luria–Bertani (LB) medium supplemented with 100 μg·ml-1 ampicillin at 37 °C and were induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at an OD600 of 1.0. After 6 h of induction, these cells were harvested through centrifugation at 4 °C and were then stored as frozen pellets at -20 °C before enzyme purification. The pellets were resuspended in 10 ml of Tris-HCl buffer (pH 8.0), disrupted by sonication, and centrifuged at 14,000 ×g for 60 min at 4 °C to remove cell debris. The supernatant was subjected to heat treatment at 70 °C for 30 min and centrifuged at 14,000 ×g to remove all denatured proteins. The clear supernatant was then applied to an open column filled with Ni2+-NTA resin (Qiagen GmbH, Hiden, Germany) according to the instruction manual. Each purification step was verified through the enzyme assay of APase activity and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the desired fractions were pooled. The pooled fractions were concentrated to less than 0.5 ml and were then applied to a column packed with Sepharose 6 (GE Healthcare) for gel filtration chromatography, yielding homogeneous protein.
2.4. Enzyme assay
7TsAPase activity was determined by measuring the release of p-nitrophenol (pNP) from pNPP. The assay mixture contained 5 mM pNPP (substrate), 1 mM MnCl2, 0.1 M Tris-HCl buffer (pH 10.5), and an appropriate amount of enzyme in a total volume of 1 ml. The reaction was initiated by adding the enzyme and was allowed to proceed at the desired temperatures. The produced pNP was found to have absorbance at 405 nm. TsAPase activity was determined from the amount of generated pNP, which was proportional to the absorbance increase at 405 nm that was measured on a spectrophotometer (JASCO V-630, Japan). One unit (U) of TsAPase activity was defined as the amount of enzyme that produced 1 micromole of pNP per min. The protein concentration was determined using the Bradford method, for which the Bio-Rad protein assay reagent was employed. Bovine serum albumin was used as the standard.
2.5. Determination of biochemical and biophysical properties
8The effect of buffer, glycerol, NaCl, and SDS on enzyme activity was determined to establish the optimal conditions for TsAPase assays. Three buffers, Tris-HCl, glycine-NaOH, and HEPES, were employed to investigate the effect of buffer on enzyme activity. To examine the effect of the chemicals on TsAPase activity, varying concentrations of glycerol, SDS, and NaCl in the standard assay solution in Tris-HCl buffer (pH 10.5) were added to the reaction. The enzyme reactions were initiated by adding 5 mM pNPP as the substrate and were continuously monitored for 3 min at 70 °C by using a spectrophotometer (JASCO V-630, Japan) at 405 nm. In all tests, relative activity was used to establish the optimal conditions for enzyme assay, and the highest activity was defined as a relative activity of 100%.
2.6. Determination of kinetic parameters
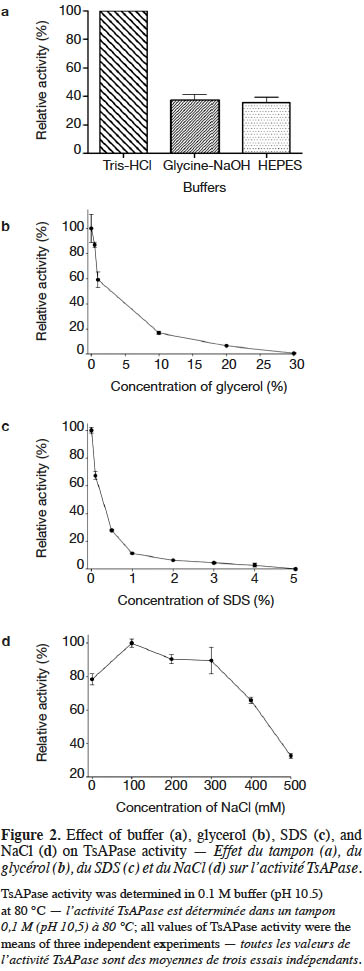
9TsAPase activity was determined by adding 20 µl of purified TsAPase (1.5 U·ml-1) in assay buffer containing 1 mM MnCl2 and 0.1 M Tris-HCl buffer (pH 10.5). The enzyme reactions were initiated by adding various concentrations of the pNPP substrate (0.01–1.0 mM). Activity was continuously monitored for 3 min at 70 °C by using a spectrophotometer (JASCO V-630, Japan) at 405 nm. Measurement was repeated three times. The kinetic parameters Km, Vmax, and kcat were determined using a Lineweaver–Burk plot (equation 1).
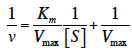
2.7. Determination of thermal properties
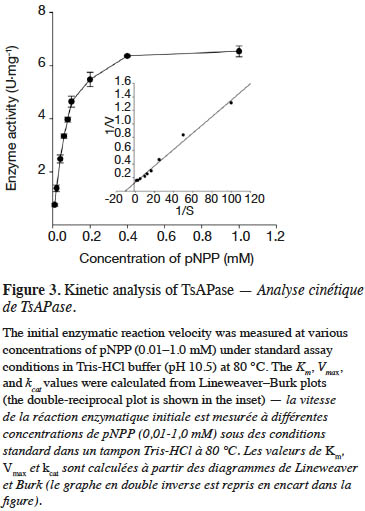
10The effect of temperature on enzyme activity was investigated in this study. To determine the optimal temperature of TsAPase, enzyme activity was examined at temperatures from 40 to 100 °C by using 5 mM pNPP as the substrate under optimal assay conditions (0.1 M Tris-HCl buffer, pH 10.5). The enzyme reactions were initiated by adding the pNPP substrate and were continuously monitored for 3 min at 70 °C by using a spectrophotometer (JASCO V-630, Japan) at 405 nm.
11To investigate the stability of the enzyme, TsAPase was stored at 4, 25, and 37 °C for 30 days. Enzyme activity was measured every day under optimal assay conditions to evaluate the stability and half-life of TsAPase at different storage temperatures.
2.8. Sequence alignment analysis
12Additional APase sequences were obtained from UniProt (http://www.uniprot.org/). These sequences were aligned using the Clustal algorithm and the BLOSUM sequence substitution table in the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/). Aligned sequences were viewed and edited using the GeneDoc program (http://iubio.bio.indiana.edu/soft/molbio/ibmpc/genedoc-readme.html).
3. Results
3.1. Expression and purification of TsAPase
13In this study, the TsAPase gene was successfully amplified from the genomic DNA of Thermus sp. NTU-237 by using PCR with primers designed according to the sequence deposited in GenBank (Accession No. KX267839). The amplified TsAPase gene fragment was then cloned into the expression vector of pQE30 to construct pQE-TsAPase. Escherichia coli cells harboring the pQE-TsAPase plasmid were cultured overnight, subcultured, and then induced with 1 mM IPTG for TsAPase expression. After induction for 6 h, the bacterial cells were harvested through centrifugation. Protein purification was performed by resuspending pellets, disrupting pellets through sonication, applying heat treatment, and conducting Ni-chelating chromatography of the supernatant and gel filtration chromatography of pooled fractions. Each purification step was verified through 12% SDS-PAGE and an assay of phosphatase activity, and the desired fraction was collected. The purified TsAPase showed more than 99% homogeneity. SDS-PAGE analysis of the purified TsAPase yielded a single band with an apparent molecular mass of 54 kDa (Figure 1a). Analysis through gel filtration chromatography revealed that TsAPase has a molecular mass of approximately 109 kDa, which indicated that TsAPase has a dimeric structure in the native form.
14To investigate the chromogenic ability of TsAPase, the purified TsAPase was also resolved through native PAGE for active staining analysis. The results showed that TsAPase catalyzed the dephosphorylation of BCIP and elicited blue color development by the BCIP/NBT reaction kit at room temperature (Figure 1b). The minimal amount of TsAPase required for blue color development was estimated to be 15 ng per well. This observation suggested that TsAPase can be applied for nonradioactive detection in clinical and basic research.

3.2. Biochemical and biophysical properties of TsAPase
15In this study, TsAPase was characterized to establish the optimal conditions for TsAPase assays. TsAPase showed the highest activity at pH10.5 in a preliminary study (Chiang, 2010). Therefore, three buffers (Tris-HCl, glycine-NaOH, and HEPES) were prepared and were added to the reaction mixture at pH10.5 at a concentration of 0.1 M. Their effects on TsAPase activity were studied. As shown in figure 2a, TsAPase showed the highest activity in Tris-HCl buffer, followed by glycine-NaOH and HEPES, with respective relative activities of 100%, 37.56%, and 35.60%.
16In general, commercial enzymes can be supplied in three forms at a low temperature: lyophilized dry powder, precipitate suspension liquid, and glycerol-containing liquid. For storing and transporting enzymes at −20 °C, glycerol is commonly used as an antifreezing agent to convert the frozen enzyme into a liquid form. To determine the effect of glycerol on TsAPase activity, various concentrations of glycerol (0%–30%) were added to the assay reaction. The results showed that glycerol had an inhibitory effect on TsAPase activity. The addition of a higher concentration of glycerol resulted in lower TsAPase activity (Figure 2b).
17Figure 2c presents the effect of SDS on TsAPase activity. Similar to glycerol, SDS inhibited enzyme activity. SDS significantly decreased TsAPase activity, and 5% SDS fully inactivated TsAPase. In contrast to SDS and glycerol, NaCl at concentrations between 100 and 300 mM increased enzyme activity, as shown in figure 2d. The highest enzyme activity was observed when 100 mM NaCl was added to the assay reaction, and enzyme activity was inhibited and observed to decrease when high concentrations of NaCl (300 mM or higher) were added to the assay reactions.
3.3. Kinetic parameters of TsAPase
18The kinetic parameters of TsAPase were determined by measuring the release of pNP from pNPP under optimal conditions. After monitoring the absorbance, data were collected to calculate kinetic parameters from a Lineweaver–Burk plot. The results showed that the Km, kcat, and Vmax values of TsAPase were 81 µM, 6.08 s-1, and 6.76 U·mg-1, respectively (Figure 3).
3.4. Thermal properties of TsAPase
19TsAPase has been reported to be stable at high temperatures (Chiang, 2010). In this study, under optimal conditions, the effect of temperature on TsAPase activity was investigated from 40 to 100 °C. The results indicated that TsAPase exhibited maximum activity at 80 °C, with a specific activity of 10.68 U·mg-1 (Figure 4). In addition, at the temperatures of 90 and 100 °C, TsAPase retained more than 50% of its activity.
20Figure 5 illustrates the stability analysis of TsAPase. The storage of TsAPase at 4, 25, and 37°C for 30 days resulted in a loss of 58%, 68%, and 70% of its activity, respectively. The results also showed that TsAPase retained 50% of its activity when stored for 15 days at the three temperatures of 4, 25, and 37 °C (Figure 5). This observation suggested that TsAPase is considerably stable at room temperature.
3.5. Sequence alignment analyses
21Comparison of the translated peptide sequence of TsAPase with APases from different sources showed a significant difference (Figure 6). However, the residues (numbered by matured E. coli APase) in the active site, including S102, H331, H412, D327, D51, D369, H370, T155, and E322, were highly conserved among all APases, regardless of whether they were from thermophiles or other species (Figure 6). This result indicated that the catalytic mechanism of these APases is the same as that of other APases.

4. Discussion
22APases have been reported to have monomeric, dimeric, or multimeric structures. Of these structures, the dimeric structure is the most common among all APases (Ásgeirsson & Andrésson, 2001). In addition, APases from different sources exhibit different properties, including molecular mass, catalytic activity, kinetic parameters, and optimum pH. For example, the molecular mass of mammalian APases ranges from 130 to 170 kDa (Cho-Ngwa et al., 2007), whereas APase from E. coli consists of two identical subunits with a molecular mass of 47 kDa (Zappa et al., 2004). In this study, recombinant TsAPase was identified as having a dimeric structure in the native form and a molecular mass of 109 kDa.
23In this study, kinetic analysis revealed that the Km and kcat values of TsAPase were 81 µM and 6.08 s-1, respectively. These results are slightly different from those of previous studies. For example, the respective Km values of APases from Thermus thermophilus XM, Thermus caldophilus, and Meiothermus ruber were reported to be 34 µM, 36 µM, and 55 µM (Li et al., 2007). The Km and kcat values of APase from Thermus yunnanensis were reported to be 62 µM and 76 s-1, respectively (Gong et al., 2005). In addition, the Km and Vmax values of APases from Pyrococcus abyssi were 166.33 µM and 4.07 U·mg-1, respectively (Zappa et al., 2001). These kinetic parameters might slightly differ because APases from different sources possess different properties.
24The optimal conditions for TsAPase assays were established by investigating the effects of glycerol, SDS, and NaCl on enzyme activity. The results showed that glycerol inhibited TsAPase activity. The inhibitory effect may be due to the effect of glycerol on the ability of the enzyme to dissolve in a solution, because glycerol may form hydrogen bonds with surrounding water. The inhibitory effect may also be due to the interaction of TsAPase with glycerol; this interaction may result in some structural changes or interference with substrate binding, leading to decreased enzyme activity. This finding suggests that the reaction mixture should not contain glycerol. The effect of the anionic detergent SDS on TsAPase activity was also examined. Similar to glycerol, SDS strongly inhibited enzyme activity at a concentration of 5%. Moreover, TsAPase activity was affected by the ionic strength. The optimal concentration of NaCl was found to be 100 mM, which increased enzyme activity 1.27-fold (Figure 2d). At a concentration of 400 mM, NaCl slightly decreased TsAPase activity. This result suggested that a low concentration of NaCl is appropriate for TsAPase, whereas NaCl at a high concentration may disrupt hydrogen bonding or bind to the active site, leading to the loss of enzyme activity.
25The optimal temperature and pH of TsAPase vary among species. In this study, the optimal temperature for maximum enzyme activity was identified to be 80 °C. The TsAPase investigated in this study is more favorable than other reported TsAPases, because it retained its activity at a high temperature. The optimal pH and temperature of APase from T. thermophilus were reported to be 12 and 75 °C, respectively (Li et al., 2007). TsAPase from Bacillus stearothermophilus showed the highest activity at pH 9.0 and 60–70 °C (Mori et al., 1999). Recently, thermostable APase from Bacillus licheniformis was reported to exhibit the highest activity at 50 °C (Divya et al., 2016). Moreover, our TsAPase was stable at room temperature and could retain its activity for a long time, with a half-life of 10 days at 37 °C. Our TsAPase showed high thermostability, an important characteristic for the industrial applications of APase. Our highly thermostable enzyme may be useful for new applications of APase at a high temperature.
26To clarify the catalytic mechanism and high activity of TsAPase, the translated peptide sequence of TsAPase was compared with that of APases from other sources. Residue S102 in E. coli APase is involved in the formation of the phosphoseryl intermediate; this residue was conserved in TsAPase. Residues H331, H412, and D327 are involved in coordinating Zn12+, and residues D51, D369, and H370 are involved in coordinating Zn22+. Magnesium is bound by residues D51, T155, and E322 (Kim & Wyckoff, 1990). These eight residues (H331, H412, D327, D51, D369, H370, T155, and E322) were also conserved in TsAPase. This observation indicated that the catalytic mechanism of TsAPase is the same as that of other APases. In addition, the secondary Mg ion, which is coordinated by D153 and K328, is replaced by histidine in APases from T. thermophilus, T. caldophilus, and M. ruber. This replacement was also observed in TsAPase (Figure 6). A previous study showed that the replacement of D153 with histidine resulted in enhanced enzyme activity, because Zn2+ bound in this site was replaced by Mg2+ (Murphy et al., 1993). Therefore, this replacement might cause an increase in TsAPase activity. However, no further experimental data are available to support this notion.
27APases are widely used as nonradioactive markers or reporters for detecting special proteins or DNA targets. The only commercially available APases are obtained from bovine intestinal mucosa, porcine kidney, rabbit intestine, shrimp, and E. coli. Currently, no thermostable APase is available from commercial suppliers. TsAPase is a thermostable APase with a long shelf life and is suitable for conjugation with antibodies, target proteins, and DNA for detection under denaturing conditions. The present study showed that TsAPase possessed higher activity (10.68 U·mg-1) than APases from other thermophiles and had high stability at a high temperature. Moreover, TsAPase could catalyze the dephosphorylation of BCIP. It is an economical and environmentally safe biocatalyst for industrial applications. Our findings suggest that thermostable TsAPase is a promising marker for commercial applications in the medicine and biotechnology industries.
5. Conclusions
28TsAPase was successfully expressed in Escherichia coli and characterized. TsAPase exhibited thermostability and high catalytic efficiency. Moreover, TsAPase could catalyze the dephosphorylation of BCIP and could elicit blue color development by the BCIP/NBT reaction kit at room temperature. TsAPase shows potential for novel industrial applications.
29Acknowledgements
30This work was supported by the research grant (NSC97-2313-B-264-003-MY3) from the Ministry of Science and Technology, Taiwan, partly supported by the Promotion Project of International English-Taught Graduate Program (MOE IETGP-CCU 2014) from Ministry of Education, Taiwan, and the annually regular research fund from Chinese Culture University. H.C. Nguyen received scholarships from Chinese Culture University (Taipei, Taiwan) and supported by the regular research fund from Ton Duc Thang University (Ho Chi Minh City, Vietnam) to conduct this project. Here we thank Prof. San-San Tsay (Department of Life Sciences, National Taiwan University) for kindly providing Thermus sp. NTU-237 for this study.
Bibliographie
Ásgeirsson B. & Andrésson Ó.S., 2001. Primary structure of cold-adapted alkaline phosphatase from a Vibrio sp. as deduced from the nucleotide gene sequence. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1549, 99-111.
Banik R.M. & Pandey S.K., 2009. Selection of metal salts for alkaline phosphatase production using response surface methodology. Food Res. Int., 42, 470-475.
Barbosa J.A. et al., 2008. Purification and biochemical characterization of thermostable alkaline phosphatases produced by Rhizopus microsporus var. rhizopodiformis. Folia Microbiol., 53, 509-516.
Chiang Y.H., 2010. Cloning and characterization of thermostable alkaline phosphatase from Thermus sp. NTU-237. Master thesis: Yuanpei University, Department of Biotechnology and Pharmaceutical Technology, Hsinchu City (Taiwan).
Cho-Ngwa F., Mbua E.N., Nchamukong K.G. & Titanji V.P., 2007. Detection, purification and characterisation of a secretory alkaline phosphatase from Onchocerca species. Mol. Biochem. Parasitol., 156, 136-143.
Claussen J.C. et al., 2015. Probing the enzymatic activity of alkaline phosphatase within quantum dot bioconjugates. J. Phys. Chem. C, 119, 2208-2221.
Coleman J.E., 1992. Structure and mechanism of alkaline phosphatase. Annu. Rev. Biophys. Biomol. Struct., 21, 441-483.
Deng J., Yu P., Wang Y. & Mao L., 2015. Real-time ratiometric fluorescent assay for alkaline phosphatase activity with stimulus responsive infinite coordination polymer nanoparticles. Anal. Chem., 87, 3080-3086.
Divya A., Santhiagu A. & Prakash S.J., 2016. Cloning, expression and characterization of a highly active thermostable alkaline phosphatase from Bacillus licheniformis MTCC 1483 in Escherichia coli BL21 (DE3). Appl. Biochem. Microbiol., 52, 358-365.
Dong G. & Zeikus J.G., 1997. Purification and characterization of alkaline phosphatase from Thermotoga neapolitana. Enzyme Microb. Technol., 21, 335-340.
Guimaraes L.H.S., Terenzi H.F., Jorge J.A. & Polizeli M.L.T.M., 2001. Thermostable conidial and mycelial alkaline phosphatases from the thermophilic fungus Scytalidium thermophilum. J. Ind. Microbiol. Biotechnol., 27, 265-270.
Gong N. et al., 2005. Characterization of a thermostable alkaline phosphatase from a novel species Thermus yunnanensis sp. nov. and investigation of its cobalt activation at high temperature. Biochim. Biophys. Acta, Proteins Proteomics, 1750, 103-111.
Karamyshev A.L. et al., 1998. Processing of Escherichia coli alkaline phosphatase: role of the primary structure of the signal peptide cleavage region. J. Mol. Biol., 277, 859-870.
Kim E.E. & Wyckoff H.W., 1990. Structure of alkaline phosphatases. Clin. Chim. Acta, 186, 175-187.
Kim Y.J., Park T.S., Kim H.K. & Kwon S.T., 1997. Purification and characterization of a thermostable alkaline phosphatase produced by Thermus caldophilus GK24. J. Biochem. Mol. Biol., 30, 262-268.
Li J., Xu L. & Yang F., 2007. Expression and characterization of recombinant thermostable alkaline phosphatase from a novel thermophilic bacterium Thermus thermophilus XM. Acta Biochim. Biophys. Sin., 39, 844-850.
Liang J. et al., 2013. Fluorescent light-up probe with aggregation-induced emission characteristics for alkaline phosphatase sensing and activity study. ACS Appl. Mater. Interfaces, 5, 8784-8789.
Minamihata K., Goto M. & Kamiya N., 2012. Activation of Pyrococcus furiosus alkaline phosphatase by divalent metal ions. Biotechnol. Lett., 34, 2055-2060.
Mori S. et al., 1999. Purification and characterization of alkaline phosphatase from Bacillus stearothermophilus. Biotechnol. Appl. Biochem., 29, 235-239.
Murphy J.E., Xu X. & Kantrowitz E.R., 1993. Conversion of a magnesium binding site into a zinc binding site by a single amino acid substitution in Escherichia coli alkaline phosphatase. J. Biol. Chem., 268, 21497-21500.
Pantazaki A.A., Karagiorgas A.A., Liakopoulou-Kyriakides M. & Kyriakidis H.A., 1998. Hyperalkaline and thermostable phosphatase in Thermus thermophilus. Appl. Biochem. Biotechnol., 75, 249-259.
Peck A. et al., 2016. Tungstate as a transition state analog for catalysis by alkaline phosphatase. J. Mol. Biol., 428, 2758-2768.
Rodrigues M.C.A. et al., 2006. Acid and alkaline phosphatase activities of a fraction isolated from Parawixia bistriata spider venom. Toxicon, 47, 854-858.
Sambrook J., Fritsch E.F. & Maniatis T., 2001. Molecular cloning: a laboratory manual. 3rd ed. New York, NY, USA: Cold Spring Harbor Press.
Trowsdale J., Martin D., Bicknell D. & Campbell I., 1990. Alkaline phosphatases. Biochem. Soc. Trans., 18, 178-180.
Yuan Y.Z. et al., 1998. Thermostable alkaline phosphatase from Thermus sp. FD3041: cloning of the gene and expression in Escherichia coli. J. Genet. Genomics, 25, 375-380.
Yurchenko J.V. et al., 2003. Cloning of an alkaline phosphatase gene from the moderately thermophilic bacterium Meiothermus ruber and characterization of the recombinant enzyme. Mol. Genet. Genomics, 270, 87-93.
Zappa S. et al., 2001. Characterization of a highly thermostable alkaline phosphatase from the euryarchaeon Pyrococcus abyssi. Appl. Environ. Microbiol., 67, 4504-4511.
Zappa S., Boudrant J. & Kantrowitz E.R., 2004. Pyrococcus abyssi alkaline phosphatase: the dimer is the active form. J. Inorg. Biochem., 98, 575-581.
