


- Accueil
- Volume 22 (2018)
- Numéro 4
- Development of PCR screening assays focused on gene-coding sequences for GMO detection


Visualisation(s): 3740 (18 ULiège)
Téléchargement(s): 1125 (5 ULiège)

Development of PCR screening assays focused on gene-coding sequences for GMO detection

Notes de la rédaction
Received 16 April 2018, accepted 29 August 2018, available online 19 September 2018
This article is distributed under the terms and conditions of the CC-BY License (http://creativecommons.org/licenses/by/4.0)
Résumé
Développement de tests PCR focalisés sur les séquences codantes des gènes pour la détection des OGM
Description du sujet. Cet article présente le développement, l’évaluation et les limitations de tests de criblage basés sur des gènes et utilisés dans le cadre de la détection des OGM.
Objectifs. Le but de la recherche est de proposer de nouvelles cibles basées sur la séquence codante de gènes. Ces tests viendront rejoindre un panel d’essais PCR déjà existants de manière à pouvoir réaliser une meilleure détection des OGM.
Méthode. Des méthodes basées sur la technique de PCR en temps réel avec sondes d’hybridation ont été développées pour des gènes fréquemment utilisés dans les constructions transgéniques. Les méthodes ont été évaluées sur la base de leur spécificité et de leur sensibilité.
Résultats. Huit tests PCR ont été sélectionnés sur la base des séquences des gènes bar, pat, EPSPS, gox, gus et hsp70. Deux d’entre eux ont montré un intérêt limité suite à l’obtention de signaux positifs liés à la présence d’organismes donneurs. Ces interférences sont dues à des séquences résiduelles d’Escherichia coli dans les réactifs utilisés pour l’amplification dans le cas du test gus, tandis que pour la cible hsp70, tenant compte qu’actuellement le hsp70 est uniquement introduit dans des maïs génétiquement modifiés, le test est dénué d’intérêt car le hsp70 provient du maïs. Les tests bar, pat, EPSPS et gox ont détecté avec succès les éléments structuraux introduits dans les constructions transgéniques. Plusieurs tests ont été proposés pour le gène EPSPS de manière à couvrir les différentes versions du gène.
Conclusions. Des tests PCR pour les séquences codantes des gènes bar, pat, EPSPS et gox ont été développés et ont répondu aux critères de performance souhaités en termes de spécificité et de sensibilité. Les cibles peuvent être amplifiées avec les mêmes conditions d’amplification que celles d’autres tests visant des promoteurs et des terminateurs, et peuvent donc être utilisées en combinaison, de manière à fournir une meilleure couverture des OGM ainsi que de premières indications sur les OGM en présence.
Abstract
Description of the subject. This paper describes the development, evaluation and limitations of screening PCR assays based on genes and for use in GMO detection.
Objectives. The aim of this research is to propose new PCR assays based on gene-coding sequences that will join a panel of existing screening assays to make better GMO detection possible.
Method. Real-time PCR methods using double-dye probes were developed for genes frequently encountered in GM constructs and evaluated in terms of specificity and sensitivity.
Results. Eight real-time PCR tests were designed based on the sequences of the bar, pat, EPSPS, gox, gus and hsp70 genes. Two of them proved of limited interest due to the presence of positive signals linked to the presence of the donor organisms: residual sequences of Escherichia coli in the master mixes for the gus PCR assay and while for the hsp70 PCR test, hsp70 being only used in GM maize at present, it is useless as hsp70 originates from maize. The assays for bar, pat, EPSPS and gox were found to successfully detect the corresponding structural elements introduced in GM constructs. Several PCR assays were proposed for the EPSPS gene in order to cover the different versions of the gene.
Conclusions. Real-time PCR tests for bar, pat, EPSPS and gox were developed and met the expected performance criteria in terms of specificity and sensitivity. The targets can be amplified with the same PCR conditions as PCR assays already developed for the detection of promoters and terminators, and can be used in combination on the same PCR plate in order to provide wider coverage of GMOs and initial information concerning the GMO(s) present.
Table des matières
1. Introduction
1For the detection of genetically modified organisms (GMO), screening is the first step used to determine if a GMO or its derived products are present in food or feed. If the result is positive, additional tests must be done to identify and, where necessary, quantify the genetically modified (GM) event present. The screening must be as wide as possible to cover the different possible GM events encountered on the market.
2In this paper, we present new real-time PCR screening methods (DNA-based methods) using hybridization probes (TaqMan probes) focused on gene-coding regions. These were selected on the basis of their occurrence in GM constructs (Block et al., 2013). Detection is also possible by searching for the new proteins using immunoassays (ELISA or immuno-chromatographic test strips). These techniques are well-suited for the detection of proteins produced by the EPSPS, pat, bar and cry genes (Stave, 2002; Van den Bulcke et al., 2007). However, this approach is only suitable for raw and unprocessed products. DNA-based methods were therefore preferred in this study.
3PCR assays focused on genes can be combined with those focused on promoters and terminators (Debode et al., 2013) in order to provide screening with wider coverage. The PCR assays developed in this paper were based on the sequences of the bar, pat, EPSPS, gox, gus and hsp70 genes.
1.1. bar
4The bar gene coding for phosphinothricin acetyltransferase confers tolerance to glufosinate, an organophosphorus compound present in many non-selective systemic herbicides. Glufosinate, or its ammonium salt (glufosinate-ammonium), is the active ingredient of herbicides containing phosphinothricin (Basta, Rely, Finale, and Liberty) and acts by inhibiting glutamine synthetase, an enzyme that detoxifies ammonia by incorporation into glutamine; an accumulation of ammonia in the tissues leads to plant death. The phosphinothricin acetyltransferase catalyzes the acetylation of phosphinothricin ammonium and transforms glufosinate into an inactive compound. The bar gene was isolated from Streptomyces hygroscopicus (a soil microorganism).
5Many primers have been proposed for the detection of the bar gene by classical PCR (Ehlers et al., 1997; Matsuoka et al., 2002; James et al., 2003; Demeke & Ratnayaka, 2008; Guo et al., 2011; Kim et al., 2013). Few methods are available for real-time PCR (Grohmann et al., 2009 for a use with TaqMan probes; Barbau-Piednoir et al., 2014 for a monitoring of the amplification with SYBRGreen). This study proposes a new test for the detection of the bar gene by real-time PCR.
1.2. pat
6The pat gene has a similar function to that of the bar gene. It allows tolerance to herbicides containing glufosinate as an active ingredient. However, it comes from another organism, Streptomyces viridochromogenes, and more particularly from the TU 494 strain, which produces the phosphinothricin acetyl transferase enzyme. Primers have already been proposed for classical PCR by Matsuoka et al. (2002), Permingeat et al. (2002) and Lu et al. (2010). Due to variations in the sequence of the pat gene in different GM plants, developed primers are not always able to detect different variants of the pat gene (Lu et al., 2010). Ideally, specificity should be verified experimentally. A test for the detection of pat by real-time PCR was proposed by Weighardt et al. (2004), but is specific to T25 maize. A PCR assay allowing better coverage was developed by Zeitler et al. (2002). A new test is proposed in this study.
1.3. EPSPS
7The EPSPS gene produces the 5-enolpyruvylshikimate-3-phosphate synthase enzyme, the function of which is to synthesize aromatic amino acids (Tohidfar & Khosravi, 2015). Herbicides with glyphosate as active ingredient have a broad spectrum of action on plant species. Glyphosate acts by blocking the natural EPSPS enzyme of plants. The consequence is a lack of production of essential amino acids required for protein synthesis, which in the long run results in plant death. The additional EPSPS gene introduced in GM plants confers tolerance to glyphosate through a better distinction between glyphosate and its natural substrate (phosphoenolpyruvate or PEP). The glyphosate-tolerant EPSPS gene comes from Agrobacterium tumefaciens. The EPSPS sequence introduced into GM plants is not exactly the same as that of bacterial origin, because codon use has been adapted for plants (Chhapekar et al., 2015). This is a cause of variability that may lead to difficulties for the development of primers and probes. PCR assays relating to EPSPS detection already exist. However, these tests all seem to be focused on the junction between the chloroplastic transit peptide (CTP) and the EPSPS coding region. When the CTP2-EPSPS region is targeted, GM plants with the CTP4-EPSPS sequence, such as Roundup Ready soybean (GTS 40-3-2), will not be detected. This is because the CTP sequence can originate from either Petunia (CTP4) or Arabidopsis (CTP2), and there are significant differences between the two. Some classical PCR tests target an amplicon that is too large (Vaïtilingom et al., 1999; Demeke et al., 2002; Matsuoka et al., 2002; Wang et al., 2011; Kim et al., 2013). The effectiveness of a real-time PCR is generally better if the amplicon size is below 120 base pairs (Debode et al., 2017a). Moreover, the addition of a probe will increase the specificity of the PCR test. Real-time PCR tests have been developed by several teams (Kuribara et al., 2002; Zhang et al., 2003; Corbisier et al., 2005; Wang et al., 2011; Köppel et al., 2014). Most of these PCR tests present specificity to particular GM events. The published primers that currently offer the best coverage are those of Grohmann et al. (2009) (88 bp). They are able to detect GT73 rapeseed, MON1445 cotton, MON809, NK603 and MON88017, MON89788 soybean and GTSB77 and H7-1 sugar beet. However, the real-time PCR test of Grohmann et al. is based on the CTP2-EPSPS junction and is thus not able to detect Roundup Ready soybean (CTP4-EPSPS).
8The PCR assays proposed here only focus on the EPSPS region and not on a CTP-EPSPS junction.
1.4. gox
9The gox gene codes for a modified version of glyphosate oxidase, a bacterial enzyme derived from Ochrobactrum anthropi. The gene has been modified to improve the enzyme’s affinity towards glyphosate.
10Glyphosate oxidase (gox) accelerates the degradation of glyphosate (the active ingredient of the Roundup Ready herbicide) to aminomethylphosphonic acid (AMPA) and glyoxylate. AMPA is the primary metabolite of glyphosate and is degraded by various microorganisms, while glyoxylate is commonly found in plants and is degraded by the glyoxylate cycle, which performs the metabolic conversion of lipids. CTP4-EPSPS and glyphosate oxidase enzymes allow the plant to be more tolerant to herbicides containing glyphosate. The real-time PCR assay for gox developed in this work is based on a sequence previously identified by Matsuoka et al. (2002), who developed a classical PCR test targeting a 103 bp amplicon.
1.5. gus
11The beta-D-glucuronidase (also called gus, gusA or uidA) codes for the Escherichia coli uidA gene. This gene was introduced as a marker gene to identify transformed cells. In the presence of a suitable substrate, the gus enzyme catalyzes a colorimetric reaction which generates a blue color highlighting the transformed cells. Detection can be done by fluorimetric, spectrophotometric or histochemical methods (Cervera, 2005).
12To our knowledge, the only detection method used for uidA is a classical PCR method used in multiplex which produces an 82 bp amplicon (Randhawa et al., 2010). In this study, a real-time PCR test is proposed which generates a 71 bp amplicon. The limitations of this PCR test are also described.
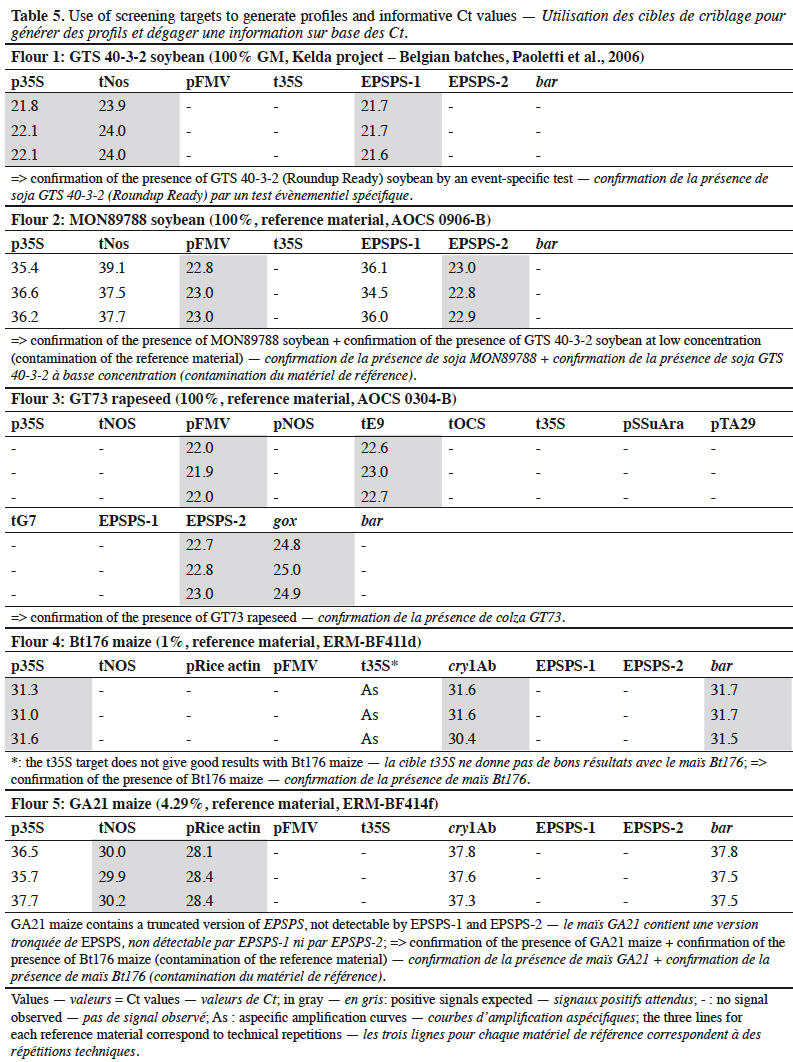
1.6. hsp70
13The intron of the hsp70 gene derived from maize has been introduced in several GM events to stabilize the transcription level of the associated gene. A PCR assay for hsp70 is presented in this paper and its limitations are discussed.
2. Materials and methods
2.1. Samples
14We obtained certified transgenic reference material (CRM) as samples, provided by the Institute for Reference Materials and Measurements (IRMM, Geel, Belgium) and the American Oil Chemists' Society (AOCS, Urbana, Illinois, USA). Plants used for specificity testing were collected in gardens or fields.
2.2. DNA extraction
15Genomic DNA was extracted and purified from all samples according to the CTAB-based method described in Annex A.3.1 of the ISO 21571 (2005) international standard. The quality and quantity of DNA extracted from samples were estimated spectrophotometrically using a Nanodrop ND-1000 spectrophotometer at 260 nm (A260) and 280 nm (A280) absorbance. DNA purity was determined using the A260/A280 ratio.
2.3. Primers and probes
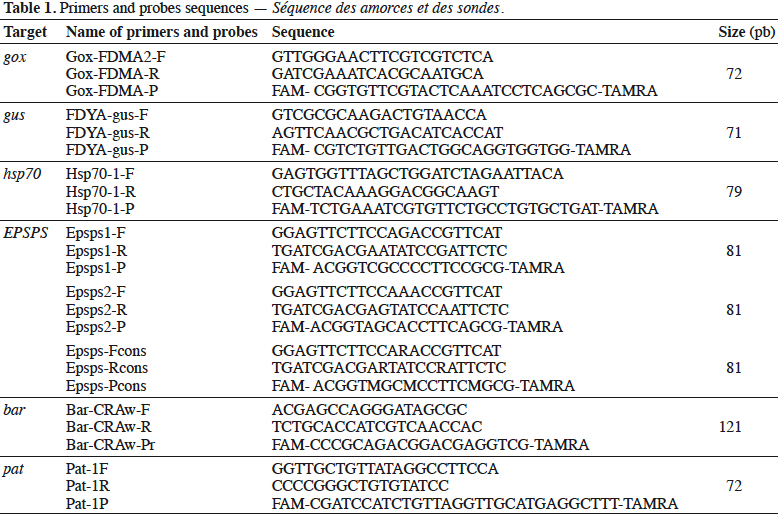
16Eurogentec (Seraing, Belgium) synthesized primers and probes. The probes were labeled with the reporter dye FAM at the 5’end, and the quencher dye TAMRA at the 3’end. The primer and probe sequences are presented in table 1.
2.4. Real-time PCR
17Real-time PCR assays (total reaction volume of 25 µl) were performed on ABI7000 and ABI7500 fast thermocyclers (Applied Biosystems, Foster City, CA) using real-time PCR master mix from Diagenode (Universal Master Mix, GMO-UN-A600, Seraing, Belgium), Eurogentec (qPCRTM Mastermix, RT-QP2X-03) or Life Technologies (TaqMan® Universal Master Mix, 4324020). The reaction mixture included 12.5 µl of master mix, 1.7 µl of each primer (5 µM), 1.5 µl of probe (9 µM), 2.6 µl of bidistilled water, and 5 µl of DNA. Reaction mixtures were distributed on 96-well reaction plates (Applied Biosystems) developed for the specific thermocyclers. The wells were covered with adhesive film, and centrifuged (500 rpm, 10 sec) to eliminate any air bubbles in the well bottoms. PCR conditions were as follows: 2 min at 50 °C; 10 min at 95 °C; 50 cycles of 15 sec at 95 °C; and 1 min at 60 °C. The baseline (normalized fluorescent signal before exponential PCR amplification occurs) was adjusted three cycles earlier than the most abundant sample, and a threshold fluorescence level was fixed in the middle of the linear region of the amplification curve represented in a semi-logarithmic graph (Y-axis with log fluorescence level as a function of PCR cycles). Between 10 and 100 ng of DNA was used for specificity testing. The amplifiability of DNA has been successfully checked with rbcL primers (Debode et al., 2012).
2.5. Cloning into pCR2.1
18The amplified fragments were introduced into the 3.9 kb pCR®2.1-TOPO plasmid vector (Invitrogen, Merelbeke, Belgium) according to the TOPO® TA Cloning® kit instructions (Invitrogen, Merelbeke, Belgium). The PCR results were visualized on 2.5% agarose gels, and the concentration was estimated by visual comparison with a quantitative molecular weight marker (Smart Ladder, Eurogentec, Seraing, Belgium). Plasmid DNA was isolated from bacterial cultures using the High Pure Plasmid Isolation kit (Roche Diagnostics, Mannheim, Germany).
2.6. Limit of detection (LOD) determination
19PCR assays sensitivity was evaluated according to the recommendations of the former AFNOR XP V03-020-2 standard (AFNOR, 2008; Broeders et al., 2014). Based on this standard, the absolute limit of detection (LOD) was determined for the PCR assay (primers + probe + amplification program) on dilutions of homozygous material (100%) or reference material.
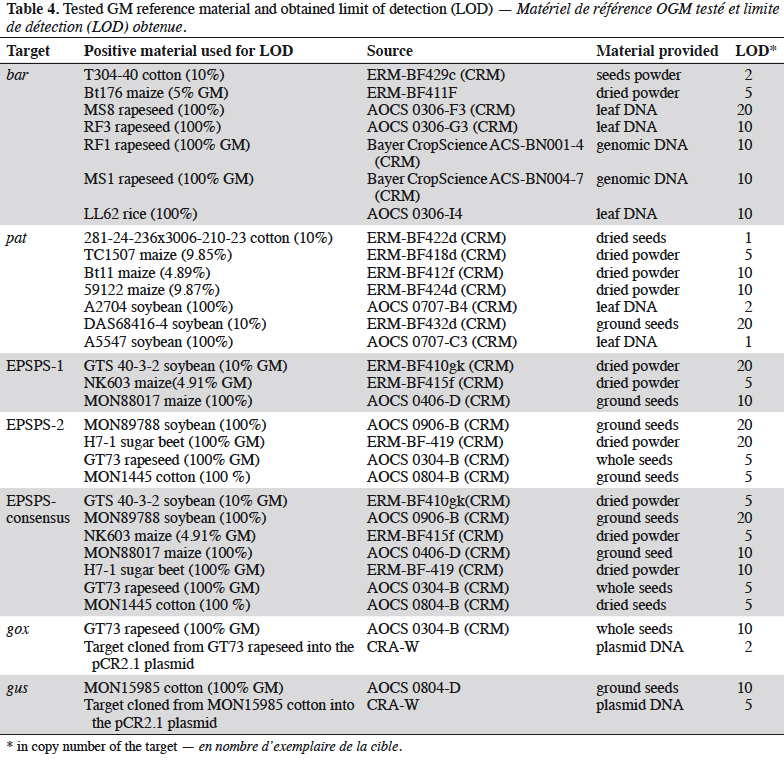
20The subsequent dilutions had to contain approximately 50, 20, 10, 5, 2, 1, and 0.1 copies of the target per reaction. Six PCRs had to be achieved for each dilution. The method’s LOD was the smallest copy number for which the six PCRs were positive, and only if PCR on the final dilution containing the 0.1 copy generated a maximum of one positive signal on the six replications. If more than one positive signal was observed for the 0.1 copy, then the DNA quantities used for the dilutions had to be checked, as this meant that they had been underestimated. The highest acceptable absolute LOD required for a test was 20 copies.
21The zygosity of some reference material was not always indicated by the provider. For the purpose of calculating the copy numbers of the target per haploid genome equivalent, such material was considered as homozygous for the transgenic trait. The measured LOD remained valid because it was expressed as “below or equal to (≤)” a copy number. For some PCR tests (gox and gus), the LOD was also checked on the cloned target. This had the advantage of providing a more accurate knowledge of the copy numbers than could be gained with certified reference material, which is generally only certified for the mass fraction.
2.7. Dilutions
22Dilutions for LOD determination were conducted in water until an estimated 20,000 copies·5 µl-1 had been reached. Further dilutions below this estimated copy number used a solution containing 5 ng·µl-1 of salmon sperm DNA as background DNA. Low-binding tubes were chosen to minimize DNA loss due to tube wall binding.
2.8. Estimated number of haploid genomes.
23The mean estimated DNA quantities necessary to obtain 20,000 target copies were based on data from Arumuganathan & Earle (1991) about sizes of haploid genome per plant species and were as follows: soybean: 23.00 ng; rapeseed: 24.60 ng; maize: 52.00 ng; sugar beet: 15.70 ng; cotton: 46.55 ng; and rice: 8.00 ng. These quantities were used to estimate the copy number of the transgenic target. Calculations were based only on Arumuganathan & Earle’s figures and not on those of other references (Bennett & Leitch, 2005; Doležel & Greilhuber, 2010; Praca-Fontes et al., 2011) because the latter do not cover all species handled in this study.
3. Results and discussion
24The several proposed methods were tested for their specificity and sensitivity, the most important criteria for evaluating the fitness for purpose of the PCR tests.
3.1. bar
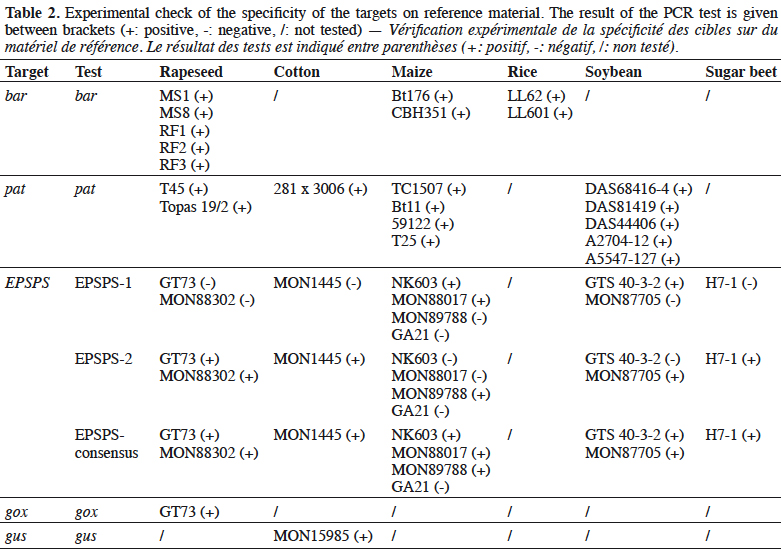
25The bar gene is present in CBH351, DAS6275, DBT418, MS3, MS6, DLL25 and Bt176 maize, LL25, T304-40, T303-3 and GHB-119 cotton, LL06, LL601 and LL62, GM II-Youming86, Zhongua91(a) and Zhongua91(b) rice, RM3-3, RM3-4, RM3-6 chicory, MS1, MS3, MS8, “PHY14, PH35”, PHY23, PHY36, RF1, RF2 and RF3 rapeseed and “W62, W98” soybean. The PCR assay developed in this study targeted a 121 bp amplicon. The ability to detect GM events containing the bar gene was positively evaluated on the available reference materials namely MS1, MS8, RF1, RF2 and RF3 oilseed rape, CBH351 and Bt176 maize and LL62 and LL601 rice (Table 2). No specificity was observed with the tested plant species (Table 3). The limit of detection was also ≤ 20 copies on the reference materials tested (Table 4). The bar PCR assay developed in this study is therefore suitable for GMO detection. However, the bar PCR assay proposed by Grohmann et al. (2009) (60 bp) also showed excellent performance for GMO detection and has been validated at international level using the pat/bar duplex (Debode et al., 2017b).
3.2. pat
26The pat gene is present in Bt10, Bt11, event 32, 676, 678, 680, T14, T25, TC1507 and DAS59122 maize, 281-24-236, 3006-210-23 and MXB-13 cotton, Falcon GS / 40/90, HCN10, Liberator L62, T45, Topas 19/2 and HCR1 rapeseed, BR891 broccoli, A2704-12, A2704-21, A5547-35, A5547-127, GU262 and DAS68416-4 soybean and T-120-7 and T252 sugarbeet. A PCR assay targeting a 72 bp amplicon was developed in this study. The ability of the assay to detect its target was positively evaluated on the available reference materials containing the pat gene, namely T45 and Topas 19/2 rapeseed, TC1507, Bt11, DAS59122 and T25 maize, 281-24-236 x 3006- 210-23 cotton, and, DAS68416-4, DAS81419, DAS44406, A2704-12 and A5547- 127 soybean (Table 2). No signal was observed with the 32 non-transgenic plant species tested (Table 3). The observed detection limit was ≤ 20 copies on the different reference materials tested (Table 4).
3.3. EPSPS
27The development of primers and probes for the detection of the EPSPS gene is complex because of the multiple versions that have been introduced in GM plants. Some GM events clearly advertise that they contain a truncated version (e.g. GA21 maize). For other GM constructs, the differences are smaller and can be localized through the alignments, but not all the EPSPS sequences of GM plants are available. It is therefore difficult to develop a single PCR assay for all the transgenic events including a glyphosate-tolerant EPSPS function. The alignment of several known sequences or portions of sequences (patents, sequences from the NCBI database) shows that two main versions of EPSPS coding regions have been introduced in plants for the production of GM events, making two PCR assays necessary: EPSPS-1 and EPSPS-2.
28In parallel we also used classical PCR to test the primers targeting the EPSPS gene mentioned in the literature, in order to assess their coverage. The primers used for comparison were those from the system developed by Eppendorf Array Technologies (Remacle & Hamels, 2006) for microarrays, those developed by the Institute of Public Health (Van Den Bulcke et al., 2014, US Patent 8,700,336) for the SYBRGreen format, the primers proposed by Matsuoka et al. (2002) for real-time PCR and the primers developed by LGL (Oberschleißheim, Germany) for detection by real-time PCR with TaqMan probes (Huber Ingrid, personal communication).
29As a result of this comparative testing, the best coverage was observed with the IPH primers. However this assay involved three primers targeting two amplicons of different sizes (94 and 124 bp), a single amplicon being insufficient to cover the different sequences. The primers developed by IPH were also the only ones that made it possible to detect the truncated EPSPS present in GA21 maize. The primers of Matsuoka et al. (2002) and those developed in this study presented the best coverage when a single pair of primers was used but they could not detect the truncated version of EPSPS found in the event GA21 (Table 2). The EPSPS-1 assay detected MON88017 and NK603 maize as well as GTS 40-3-2 soybean (Table 2). A second assay EPSPS-2 was designed to detect H7-1 sugar beet, GT73 and MON88302 rapeseed, MON87705 soybean, MON89788 maize and MON1445 cotton (Table 2). A combination of these two assays provided good coverage of the GM events containing the EPSPS gene. To check if a single assay was able to identify both groups of EPSPS sequences, a “consensus” assay was designed. This made use of degenerate nucleotide positions to allow hybridization with a larger panel of sequences. This EPSPS-consensus assay made it possible to detect the transgenic events covered by the EPSPS-1 and EPSPS-2 assays. However, the Ct values obtained with the EPSPS consensus-assay were later than those obtained with the EPSPS-1 and EPSPS-2 assays, with a delay of +/- 3 Ct. This loss of efficiency could be a problem if the analysis has to be carried out on highly processed products from which very little DNA can be recovered.
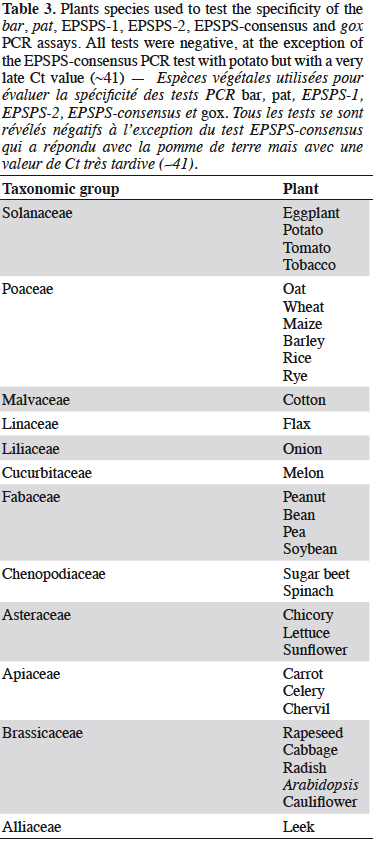
30The specificity of the EPSPS assays was tested on a panel of non-genetically modified plants (Table 3). The EPSPS-1 and EPSPS-2 assays were found to be very specific. In contrast, the EPSPS-consensus-assay gave a positive but late response (Ct of ~41) with potato. This was probably because the primers and probe used degenerate sequences.
31The EPSPS-1, EPSPS-2 and EPSPS-consensus assays were all considered to be sensitive enough with a LOD equal to or below 20 copies on the different reference materials (Table 4).
32The difference in LOD which occurred between the various reference flours may be linked to the uncertainty in the measurement of the amount of DNA present in the DNA extract, the estimated number of copies per ng referenced in the literature (Arumuganathan & Earle, 1991) and the successive dilutions to reach the low levels of copies.
3.4. gox
33The gox gene is found in GTSB77 and A5-15 sugar beet, GT73 and GT200 rapeseed, ZSR500/502 Polish canola and MON80100, MON802, MON809 and MON832 maize.
34The gox assay was developed on the basis of the sequence provided by Matsuoka et al. (2002) and generates a PCR product of 72 bp. Only reference material from GT73 rapeseed was available to test the gox target, so GT73 was used to evaluate the performance of the PCR method. The sensitivity was evaluated on genomic DNA extracted from GT73 rapeseed and plasmid DNA containing the target cloned from GT73. The PCR test did not generate unspecific signals with the 32 plant species tested that may be found in food (Table 3), and the target was well detected in GT73 rapeseed. The gox PCR assay achieved the performance criteria in terms of sensitivity with a limit of detection (LOD) below 20 copies (Table 4). The gox PCR assay was compared with another existing method (Weng et al., 2005), found in the literature after the design of the assay outlined here, that amplifies a somewhat larger target (89 bp). Similar performances were observed in terms of LOD tested on genomic DNA from GT73 rapeseed between the PCR assay of Weng et al. and the PCR assay developed in this study.
3.5. gus
35The gus gene is found in GTSB77 sugar beet, 55-1/63-1 papaya, G94-1, G94-19, G168, W62, W68, DD-026005-3 GM002-9-ACS and ACS GM001-8 soybeans, MON15985 and GTL-GFM311-7 cotton and CS plum. The only reference material currently available is MON15985 cotton.
36The primers and probes developed in this study amplify a fragment of 71 bp. The LOD of the PCR assay was tested on genomic DNA from MON15985 cotton and plasmid DNA (containing the target cloned from MON15985 cotton). A LOD of less than 20 copies was obtained on genomic and plasmid material (Table 4).
37The sequence of the target showed 100% similarity with the gus gene of Escherichia coli (results obtained with the blast function of the National Center for Biotechnology Information). Positive signals could therefore be generated by the presence of coliform bacteria in a sample.
38During the testing phase of the gus PCR assay, we noticed that positive signals were often observed in the no-template controls (purified water instead of DNA). This caused problems for specificity testing on plant species, so these data are not indicated in Table 3. The hypothesis to explain the source of these positive signals is linked to the Taq polymerases used, as they derive from cloned versions produced in Escherichia coli. The unspecific signals can be explained by the presence of DNA from Escherichia coli in the master mix. The purity may vary from one batch to another (data not shown), which would explain the presence or absence of signals in the wells supposed to be negative. The LOD was therefore determined by selecting a batch of Taq polymerase presenting weak contamination by E. coli DNA and taking into account any Ct values observed in the negative controls as cut-off values (Chandelier et al., 2010).
39This type of contamination in which the purity of Taq is implicated has already been demonstrated for the bla gene (Holst-Jensen et al., 2003), which is present in cloning vectors as a selection marker, conferring tolerance to ampicillin by its beta-lactamase activity. Due to these problems, the gus PCR assay was considered as unsuitable for routine GMO screening.
3.6. hsp70
40The intron of the hsp70 gene comes from maize and is found in the MON80100, MON802, MON809, MON810, MON832, MON89034, MON87427 and NK603 maize events. In MON15985 cotton however, hsp70 does not come from maize but from petunia, and the two sequences only have 76% similarity. Differences are distributed over the entire sequence, which impedes any development of a consensus test.
41Primers and probe were selected for the hsp70 sequence of maize and targeted an amplicon of 79 bp. This assay delivered positive signals on GM maize (MON810 and NK603 events) but also on non-GM maize due to the fact that hsp70 is naturally present in maize. We thus concluded that hsp70 is not an appropriate screening target if used on a matrix with maize. At present, however this screening element is only involved in constructs of GM maize events. The hsp70 PCR assay was therefore not considered for specificity and sensitivity testing.
42The various screening PCR methods developed in this work can be combined with other screening PCR assays focused on genes or promoters and terminators. Practical examples are presented in Table 5 for several reference flours where the PCR assays were combined with the cry1Ab gene (Debode et al., 2017c) and assays focused on promoters and terminators such as p35S and tNOS (Kuribara et al., 2002) or pFMV, pNOS, pSSuAra, pTA29, t35S, tE9, tOCS and tg7 (Debode et al., 2013). All targets were amplified with the same PCR conditions. This methodology is relevant as the EURL-GMFF aims to develop ready-to-use plates focused on screening tests (Rosa et al., 2016) capable of dealing with the arrival of new GMOs on the market. Developments combining existing screening PCR assays have already been proposed in real-time PCR using SYBRGreen (Peng et al., 2016) or double-dye probes (Scholtens et al., 2017).
43In the examples given in Table 5, the screening tests used were adapted to the analyzed plant species. It is clear that in some cases, assays may have a duplicate effect. Nevertheless, such redundant information acts as a confirmation.
44The use of a larger number of screening PCR assays will increase the cost of the screening, but will provide better coverage and may help to reduce the number of identification tests.
45The “profiles” obtained with the screening PCR tests (positive or negative signals) can be introduced in the GMOSeek matrix software (Block et al., 2013) to determine which events may be present.
46The comparison of the signal (Ct) of the different assays also makes it possible to highlight the presence of contamination or the presence of more than one event (atypical profiles, differences in signal intensities for several targets, etc.). This latter case is more difficult to handle if, for instance, two GMOs are present in similar proportions. This would necessitate an identification strategy requiring a larger number of identification tests. Examples presented in Table 5 illustrate the application of the screening PCR assays developed to generate profiles and informative Ct values. An expected profile was obtained for the soybean GTS40-3-2 with positive signals for the p35S, tNOS and EPSPS-1 PCR assays. We can however observe a difference in the Ct values between the PCR assays for the different structural elements, the tNOS PCR assay giving a later Ct, probably due to a less efficient PCR test. This later Ct for the tNOS PCR assay was also observed with the other reference materials tested.
47The MON89788 soybean gave positive signals with the pFMV and EPSPS-2 PCR assays. However a second profile was observed showing later signals for the p35S, tNOS and EPSPS-1 PCR assays. This second profile was explained by a contamination of the reference material by GTS 40-3-2 soybean. Note that reference materials are certified for the presence of the announced GM events at the defined GM percentage but this does not exclude a contamination by other events. A second profile was also observed on the reference material corresponding to the GA21 maize. The GA21 was well detected with the tNOS and pRice actin PCR assays but a second profile linked to a contamination with Bt176 maize was detected with the p35S, cry1Ab and bar assays. The identification of the GM events was realized with the event-specific tests recommended in the Compendium of reference methods for GMO analysis (ENGL, 2011).
48The GT73 rapeseed gave the expected signals with the pFMV, tE9, EPSPS-2 and gox PCR assays. The Bt176 maize gave a positive signal with the p35S, cry1Ab and bar PCR assays. Bad amplification curves were obtained with the t35S PCR assay. This was expected, as the t35S PCR assay is known to fail detection of the t35S introduced in Bt176 (Debode et al., 2013). This shows the importance that next to a matrix table listing the presence of structural elements per events, one should also consider the successfulness of each method per event.
4. Conclusions
49Six PCR assays for GMO screening, focusing on gene coding sequences frequently used in constructs, were developed and tested for use in qualitative detection. The assays for gox, EPSPS, bar and pat genes met the expected performance criteria in terms of specificity and sensitivity. The PCR assays for gus and hsp70 were not retained due to possible interfering signals if used in routine screening. These new PCR methods may be added to the panel of assays focused on promoters and terminators (e.g. Debode et al., 2013; Debode et al., 2018) or other genes such as cry1Ab (Debode et al., 2017c) in order to offer good coverage of the elements frequently encountered in transgenic constructs and to propose a system where all the targets can be amplified with similar PCR conditions. Ct values in the same range for different screening assays may indicate the possibility of a single GM event (or a mix of several GM events with the same screening elements), while important differences suggest the presence of several GM events in different concentrations. These new PCR assays may help to increase the coverage of the GM events that could be present in a sample and constitute a step forward in the detection of unauthorized/unknown GMOs.
50Acknowledgements
51This research was conducted within a Belgian research project (Convention RT-06/6 GMODetec) financed by the Belgian Federal Public Service for Public Health, Food Chain Safety, and Environment in an endeavor involving three partners (ISP/WIV, ILVO, and CRA-W).
52We are grateful to Denis Roulez, Cécile Ancion, and Gaëlle Antoine (GMO team of CRA-W) for their technical help.
Bibliographie
AFNOR XP V03-020-2, 2008. Produits alimentaires. Détection et quantification des organismes végétaux génétiquement modifiés et produits dérivés. Partie 2 : méthodes basées sur la réaction de polymérisation en chaîne. Norme expérimentale. La Plaine Saint-Denis, France : AFNOR.
Arumuganathan K. & Earle E.D., 1991. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep., 9, 208-218.
Barbau-Piednoir E. et al., 2014. Inter-laboratory testing of GMO detection by Combinatory SYBR® Green PCR Screening (CoSYPS). Food Anal. Methods, 7, 1719-1728.
Bennett M.D. & Leitch I.J., 2005. Nuclear DNA amounts in angiosperms: progress, problems and prospects. Ann. Bot., 95, 45-90.
Block A. et al., 2013. The GMOseek matrix: a decision support tool for optimizing the detection of genetically modified plants. BMC Bioinf., 14, 256.
Broeders S. et al., 2014. Guidelines for validation of qualitative real-time PCR methods. Trends Food Sci. Technol., 37, 115-126.
Cervera M., 2005. Histochemical and fluorometric assays for uidA (GUS) gene detection. In: Pena L., ed. Transgenic plants. Methods and protocols. Totowa, NJ, USA: Humana Press, 203-213.
Chandelier A., Planchon V. & Oger R., 2010. Determination of cycle cut off in real‐time PCR for the detection of regulated plant pathogens. EPPO Bull., 40, 52-58.
Chhapekar S. et al., 2015. Transgenic rice expressing a codon-modified synthetic CP4-EPSPS confers tolerance to broad-spectrum herbicide, glyphosate. Plant Cell Rep., 34, 721-731.
Corbisier P. et al., 2005. Quantitative determination of Roundup Ready soybean (Glycine max) extracted from highly processed flour. Anal. Bioanal. Chem., 383, 282-290.
Debode F., Janssen É., Marien A. & Berben G., 2012. DNA detection by conventional and real-time PCR after extraction from vegetable oils. J. Am. Oil Chem. Soc., 89, 1249-1257.
Debode F., Janssen É. & Berben G., 2013. Development of 10 new screening PCR assays for GMO detection targeting promoters (pFMV, pNOS, pSSuAra, pTA29, pUbi, pRice actin) and terminators (t35S, tE9, tOCS, tg7). Eur. Food Res. Technol., 236, 659-669.
Debode F. et al., 2017a. Influence of the amplicon length on real-time PCR results. Biotechnol. Agron. Soc. Environ., 21, 3-11.
Debode F. et al., 2017b. Inter-laboratory studies for the validation of two singleplex (tE9 and pea lectin) and one duplex (pat/bar) real-time PCR methods for GMO detection. Food Control, 73, 452-461.
Debode F., Janssen É., Bragard C. & Berben G., 2017c. Detection by real-time PCR and pyrosequencing of the cry1Ab and cry1Ac genes introduced in GM constructions. Food Addit. Contam. Part A., 34, 1398-1409.
Debode F. et al., 2018. Development of real-time PCR assays for the detection of the pin II terminator (tpinII) used in GM constructs and its donor organism, potato (Solanum tuberosum). Food Anal. Methods, 11(8), 2172-2180.
Demeke T., Giroux R.W., Reitmeier S. & Simon S.L., 2002. Development of a polymerase chain reaction assay for detection of three canola transgenes. J. Am. Oil Chem. Soc., 79, 1015-1019.
Demeke T. & Ratnayaka I., 2008. Multiplex qualitative PCR assay for identification of genetically modified canola events and real-time event-specific PCR assay for quantification of the GT73 canola event. Food Control, 19, 893-897.
Doležel J. & Greilhuber J., 2010. Nuclear genome size: are we getting closer? Cytometry A, 77, 635-642.
Ehlers B. et al., 1997. Nachweis gentechnischer Veränderungen in Mais mittels PCR. Bundesgesundheitsblatt, 40, 118-121.
ENGL (European Network of GMO Laboratories), 2011. Compendium of reference methods for GMO analysis. JRC reference reports. Luxembourg: Publications Office of the European Union.
Grohmann L., Brünen-Nieweler C., Nemeth A. & Waiblinger H.U., 2009. Collaborative trial validation studies of real-time PCR-based GMO screening methods for detection of the bar gene and the ctp2-cp4epsps construct. J. Agric. Food Chem., 57, 8913-8920.
Guo J. et al., 2011. MPIC: a high-throughput analytical method for multiple DNA targets. Anal. Chem., 83, 1579-1586.
Holst-Jensen A., Rønning S.B., Løvseth A. & Berdal K.G., 2003. PCR technology for screening and quantification of genetically modified organisms (GMOs). Anal. Bioanal. Chem., 375, 985-993.
ISO 21571, 2005. Foodstuffs. Methods of analysis for the detection of genetically modified organisms and derived products. Nucleic acid extraction. Genova, Italy: International Organization for Standardization.
James D. et al., 2003. Reliable detection and identification of genetically modified maize, soybean, and canola by multiplex PCR analysis. J. Agric. Food Chem., 51, 5829-5834
Kim J.H., Kim E.H., Yea M.C. & Kim H.Y., 2013. Validation of A multiplex PCR detection kit for screening of herbicide-tolerant genes in genetically modified crops. J. Korean Soc. Appl. Biol. Chem., 56, 251-254.
Köppel R., Sendic A. & Waiblinger H.U., 2014. Two quantitative multiplex real-time PCR systems for the efficient GMO screening of food products. Eur. Food Res. Technol., 239, 653-659.
Kuribara H. et al., 2002. Novel reference molecules for quantitation of genetically modified maize and soybean. J. AOAC Int., 85, 1077-1089.
Lu I.J., Lin C.H. & Pan T.M., 2010. Establishment of a system based on universal multiplex-PCR for screening genetically modified crops. Anal. Bioanal. Chem., 396, 2055-2064.
Matsuoka T. et al., 2002. Detection of recombinant DNA segments introduced to genetically modified maize (Zea mays). J. Agric. Food Chem., 50, 2100-2109.
Paoletti C. et al., 2006. Kernel lot distribution assessment (KeLDA): a study on the distribution of GMO in large soybean shipments. Eur. Food Res. Technol., 224, 129-139.
Peng C. et al., 2016. Development of a qualitative real-time PCR method to detect 19 targets for identification of genetically modified organisms. Springerplus, 5, 889.
Permingeat H.R., Reggiardo M.I. & Vallejos R.H., 2002. Detection and quantification of transgenes in grains by multiplex and real-time PCR. J. Agric. Food Chem., 50, 4431-4436.
Praca-Fontes M.M., Carvalho C.R., Clarindo W.R. & Cruz C.D., 2011. Revisiting the DNA C-values of the genome size-standards used in plant flow cytometry to choose the “best primary standards”. Plant Cell Rep., 30, 1183-1191.
Randhawa G.J., Chhabra R. & Singh M., 2010. Decaplex and real-time PCR based detection of MON531 and MON15985 Bt cotton events. J. Agric. Food Chem., 58, 9875-9881.
Remacle J. & Hamels S., 2006. Identification and/or quantification method of nucleotide sequence(s) elements specific of genetically modified plants on arrays. U.S. Patent Application 11/435,553.
Rosa S.F. et al., 2016. Development and applicability of a ready-to-use PCR system for GMO screening. Food Chem., 201, 110-119.
Scholtens I.M. et al., 2017. Semiautomated TaqMan PCR screening of GMO labelled samples for (unauthorised) GMOs. Anal. Bioanal. Chem., 409, 3877-3889.
Stave J.W., 2002. Protein immunoassay methods for detection of biotech crops: applications, limitations, and practical considerations. J. AOAC Int., 85, 780-786.
Tohidfar M. & Khosravi S., 2015. Transgenic crops with an improved resistance to biotic stresses. A review. Biotechnol. Agron. Soc. Environ., 19, 62-70.
Vaïtilingom M., Pijnenburg H., Gendre F. & Brignon P., 1999. Real-time quantitative PCR detection of genetically modified Maximizer maize and Roundup Ready soybean in some representative foods. J. Agric. Food Chem., 47, 5261-5266
Van den BulckeM. et al., 2007. Detection of genetically modified plant products by protein strip testing: an evaluation of real-life samples. Eur. Food Res. Technol., 225, 49-57.
Van Den Bulcke M.H.G. et al., 2014. Transgenic plant event detection. U.S. Patent No. 8,700,336. Washington: U.S. Patent and Trademark Office.
Wang X. et al., 2011. Construction of a reference plasmid molecule containing eight targets for the detection of genetically modified crops. Appl. Microbiol. Biotechnol., 90, 721-731
Weighardt F. et al., 2004. Real-time polymerase chain reaction-based approach for quantification of the pat gene in the T25 Zea mays event. J. AOAC Int., 87, 1342-1355.
Weng H. et al., 2005. Novel reference gene, high-mobility-group protein I/Y, used in qualitative and real-time quantitative polymerase chain reaction detection of transgenic rapeseed cultivars. J. AOAC Int., 88, 577-584.
Zeitler R., Pietsch K. & Waiblinger H.U., 2002. Validation of real-time PCR methods for the quantification of transgenic contaminations in rape seed. Eur. Food Res. Technol., 214, 346-351.
Zhang Y. et al., 2003. A novel real‐time quantitative PCR method using attached universal template probe. Nucleic Acids Res., 31, e123-e123.
