


- Accueil
- volume 12 (2008)
- numéro 2
- Implication des modifications épigénétiques dans les cancers : développement de nouvelles approches thérapeutiques


Visualisation(s): 10880 (61 ULiège)
Téléchargement(s): 211 (3 ULiège)

Implication des modifications épigénétiques dans les cancers : développement de nouvelles approches thérapeutiques

Notes de la rédaction
Reçu le 9 novembre 2007, accepté le 15 janvier 2008.
Résumé
Les cancers étant la seconde cause de décès après les maladies cardiovasculaires dans les pays industrialisés, il est urgent d'établir de nouvelles approches thérapeutiques. Hormis des mutations directes de l'ADN de gènes essentiels, l'expansion d'une cellule cancéreuse est fréquemment associée à des modifications épigénétiques c'est-à-dire non directement codées par la séquence de l'ADN. Parmi ces modifications épigénétiques, l'acétylation des histones et la méthylation de l'ADN semblent jouer un rôle important. Dans ce contexte, une stratégie thérapeutique actuellement très prometteuse consiste à corriger des erreurs épigénétiques à l'aide de composés modulant l'acétylation des histones et la méthylation de l'ADN utilisés seuls ou en combinaison avec d'autres agents anticancéreux.
Abstract
Involvement of epigenetic modifications in cancers: development of new therapeutic approaches. Since cancer is the second cause of death after cardiovascular diseases in industrialized countries, it is urgent to elaborate new therapeutic approaches. Besides DNA mutations of essential genes, expansion of a cancer cell is frequently associated with epigenetic modifications i.e. not directly coded by the DNA sequence. Amongst epigenetic modifications, histones acetylation and DNA methylation are known to play important roles. In this context, a very promising anticancer therapy would be to correct epigenetic errors using compounds modulating histone acetylation and DNA methylation alone or in combination with other chemotherapeutic agents.
Table des matières
1. Structure du génome
1Le génome humain se présente sous la forme d'une double hélice d'ADN (Watson et al., 1953) contenant environ 30 000 gènes et dont la taille à l'état déroulé est de deux mètres. La fibre d'ADN est organisée sous forme d'une structure appelée chromatine. Cette chromatine est composée d'ADN associé à des protéines. L'ADN est constitué d'une succession de nucléotides composés d'un sucre (le désoxyribose), d'un acide phosphorique et d'une base azotée. Il existe quatre bases azotées divisées en deux grandes familles : les bases puriques (adénine et guanine) et les bases pyrimidiques (thymine et cytosine). Dans la double hélice d'ADN, l'adénine est toujours associée à la thymine et la cytosine l'est toujours à la guanine.
2On distingue deux types de protéines liées à l'ADN : les histones et les protéines chromosomiques non histones. Les histones sont les protéines les plus abondantes de la chromatine. Elles possèdent une masse moléculaire comprise entre 10 et 24 kDa, elles sont très basiques, riches en résidus lysine et arginine et sont très conservées au cours de l'évolution (sauf pour l'histone H1). Les histones sont retrouvées uniquement chez les eucaryotes. Il en existe cinq types qui sont présents en quantités plus ou moins égales et qui se répartissent en deux familles : les histones nucléosomiques (H2A, H2B, H3 et H4) qui sont responsables de l'enroulement de l'ADN dans les nucléosomes et les histones H1 qui sont des histones internucléosomiques. Dans le noyau, la chromatine est formée d'une répétition de sous-unités appelées nucléosomes (Kornberg, 1974 ; Kornberg et al., 1995 ; Luger et al., 1997). Celui-ci est constitué de 146 pb d'ADN enroulées deux fois autour d'un octamère d'histones. Chaque octamère comprend deux exemplaires des histones H2A, H2B, H3 et H4. Deux nucléosomes sont séparés l'un de l'autre par une cinquantaine de paires de base d'ADN nu et par l'histone internucléosomique H1. Les nucléosomes vont ensuite être assemblés en des structures de plus en plus complexes comme des spirales, des solénoïdes ou des hélices pour finalement atteindre le niveau d'organisation maximal : le chromosome (Figure 1). Ces niveaux de compaction mettent notamment en jeu une famille de protéines non-histones dont la plus importante est la topo-isomérase II.
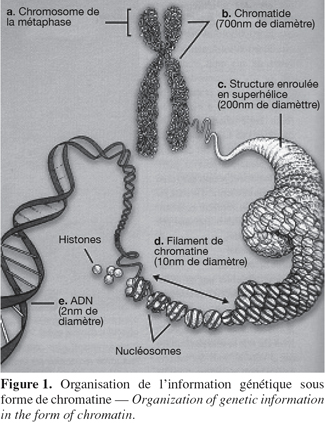
3Il existe deux types de chromatine : l'euchromatine et l'hétérochromatine. L'euchromatine correspond à la chromatine à l'état relâché alors que l'hétérochromatine correspond à des régions du génome qui sont condensées et donc inaccessibles aux facteurs de transcription (par exemple les centromères et les télomères). De cette manière, seule l'euchromatine peut être transcrite (Felsenfeld et al., 2003).
2. Les modifications post-traductionnelles des histones
4Les histones possèdent un domaine globulaire carboxy-terminal formant le cœur du nucléosome et une queue amino-terminale exposée à la surface du nucléosome (Uberbacher et al., 1985) et sujette à diverses modifications post-traductionnelles (Figure 2). Ces modifications vont influencer l'état de compaction de la chromatine et donc modifier les niveaux d'expression des gènes.
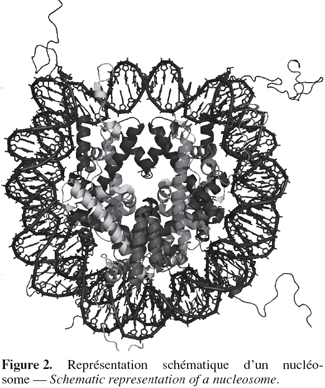
5Il existe quatre types majeurs de modification des histones : l'acétylation, la méthylation, la phosphorylation et l'ubiquitination (Strahl et al., 2000 ; Berger, 2002). Les différentes combinaisons de ces modifications forment un code épigénétique qui, une fois lu par des domaines protéiques spécifiques, entraîne diverses réponses à des endroits précis du génome. Il existe donc des interdépendances entre ces différentes modifications (Lacoste et al., 2003). L'acétylation des histones ainsi que la méthylation des histones ou de l'ADN sont les modifications post-traductionnelles les plus directement liées à l'état transcriptionnel de la région de l'ADN impliquée.
3. L'acétylation des histones
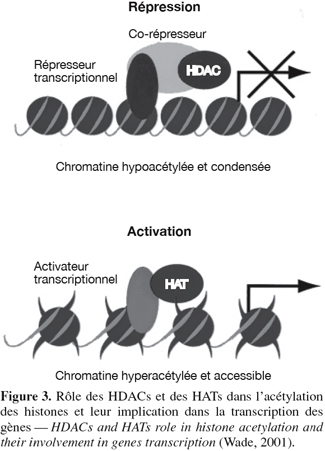
6L'acétylation est le transfert d'un groupement acétyle provenant de l'acétyle-coenzyme A sur le groupement ε-amino de certains résidus lysine. Toutes les histones sont susceptibles d'être acétylées sur des lysines spécifiques dans leur domaine amino-terminal. La neutralisation de la charge positive du groupement NH3+ de la lysine peut mener à la diminution des contacts avec l'ADN nucléosomique (possède une charge négative à cause des groupements PO43-), avec les nucléosomes voisins et avec certaines protéines régulatrices stabilisant la chromatine. Ces modifications produisent une chromatine plus flexible, où l'ADN est plus accessible. Deux familles d'enzymes influencent les niveaux d'acétylation des histones (Grabher et al., 1994 ; Kuo et al., 1998) : les histones acétyltransférases (HATs) qui ont comme rôle d'ajouter des groupements acétyles aux lysines des histones et les histones désacétylases (HDACs) qui ont un rôle contraire aux HATs, c'est-à-dire qu'elles enlèvent les groupements acétyles des histones (Figure 3). Ce phénomène, au final, donne lieu à une hypoacétylation qui mène à la répression de la transcription des gènes (Humphrey et al., 2001).
7Le domaine catalytique des HDACs est composé d'environ 400 acides aminés. Ces enzymes font partie de la superfamille des métalloenzymes à zinc et sont classées en fonction de leur homologie avec les HDACs de la levure Saccharomyces cerevisiae (de Ruijter et al., 2003). On distingue actuellement 18 HDACs humaines se répartissant en trois grandes classes distinctes (Gray et al., 2001 ; Drummond et al., 2005) :
8– Les HDACs de classe I : il s'agit des HDACs 1, 2, 3 et 8. Ces HDACs sont des homologues du répresseur transcriptionnel de levures yRPD3 (yeast Reduced Potassium Dependency 3). Ces enzymes sont exprimées dans la plupart des tissus et sont majoritairement présentes dans le noyau.
9– Les HDACs de classe II : il s'agit des HDACs 4, 5, 6, 7, 9 et 10. Ce sont des homologues de yHDA1 (yeast Histone Deacetylase-A1). Elles sont tissu-spécifiques et peuvent passer du noyau au cytoplasme et vice-versa en réponse à divers signaux cellulaires. Elles sont aussi parfois subdivisées en deux sous-classes : HDACs IIa (HDACs 4, 5, 7 et 9) et HDACs IIb (HDACs 6 et 10).
10– Les HDACs de classe III : elles ne montrent aucune similitude avec les HDACs de classe I et II. Ce sont des HDACs similaires à ySIR2 (yeast Silent Information Regulator 2) dont l'activité enzymatique dépend de NAD+. On y retrouve SIRT 1, 2, 3, 4, 5, 6 et 7. Cette classe d'HDACs est la moins bien caractérisée.
11La dernière désacétylase connue est HDAC 11. Celle-ci possède des homologies à la fois avec les HDACs de classe I et avec celles de classe II. Pour cette raison, elle n'est classée dans aucune des différentes classes.
12Les histones peuvent également subir d'autres modifications telles que la méthylation des lysines ou des arginines situées sur les extrémités amino-terminales des histones H3 et H4. Elle est assurée par les histones méthyltransférases (HMTs). La méthylation de lysines spécifiques joue souvent un rôle dans la formation de l'hétérochromatine. De son côté, la méthylation des arginines semble avoir un effet positif sur la transcription (Zhang et al., 2001). La phosphorylation est également une des modifications post-traductionnelles des histones. Il s'agit de la substitution d'un groupement hydroxyle par un groupement phosphate. Elle est réalisée par le biais de kinases sur les résidus sérine, thréonine et tyrosine. La réaction inverse est assurée par les phosphatases. La phosphorylation joue un rôle dans l'activation transcriptionnelle, dans la condensation des chromosomes mitotiques et dans la réparation de l'ADN (Redon et al., 2002 ; Ahmad et al., 2002). La dernière modification importante des histones est l'ubiquitination. Elle est habituellement liée à la dégradation de la protéine par le protéasome. Dans le cas des histones, il s'agit d'une mono-ubiquitination non dégradative d'une lysine dans le domaine carboxy-terminal des histones H2A ou H2B. Cette modification exercerait aussi une fonction dans la formation de l'hétérochromatine et la régulation de la transcription. Les enzymes responsables sont les ubiquitine ligases (Jason et al., 2002). De plus, il existe des interdépendances entre ces différentes modifications. Par exemple, la phosphorylation de la sérine 10 de l'histone H3 entraîne une acétylation plus importante de la lysine 14. Ces deux modifications répriment alors la méthylation de la lysine 9 (Berger, 2002). De même, la méthylation de l'arginine 3 de l'histone H4 augmente l'acétylation des lysines 8 et 12. En revanche, l'acétylation des résidus 5, 8, 12 et 16 inhibe la méthylation de l'arginine 3 (Zhang et al., 2001).
4. La méthylation de l'ADN
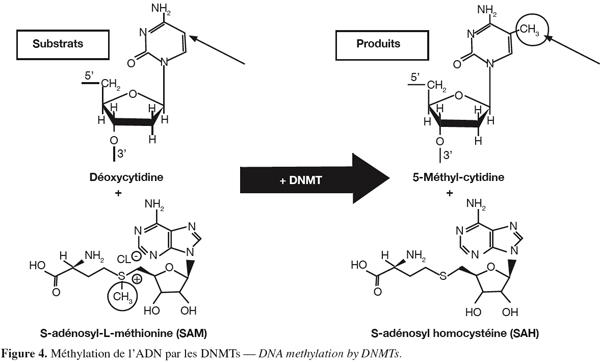
13La méthylation de l'ADN se produit principalement dans des régions appelées îlots CpG (Yoder et al., 1997). Environ 80 % de ces régions sont méthylées dans le génome humain (Cheung et al., 2005). Les DNA méthyltransférases (DNMTs) catalysent l'addition d'un groupement méthyle provenant du donneur S-adénosylméthionine sur le carbone 5 d'une cytosine (Figure 4). Parmi les DNA méthyltransférases, la DNMT1 qui est une enzyme impliquée dans le maintien de l'information épigénétique (Leonhardt et al., 1992 ; Liu et al., 1998), prend comme modèle le brin d'ADN hémi-méthylé lors de la réplication tandis que les enzymes DNMT3a et 3b peuvent agir sur un brin non méthylé et donc modifier l'information épigénétique (Okano et al., 1999). Par contre, DNMT2 ne montre pas d'activité DNA méthyltransférase. La méthylation de l'ADN conduit à la répression de l'expression du gène (Bird, 2002). Il existe aussi une enzyme (peptidyl arginine deiminase 4) pouvant, en déiminant les résidus arginine de l'histone H3, interférer avec la méthylation (Cuthbert et al., 2004). Il faut aussi savoir qu'il existe une interdépendance entre l'acétylation des histones et la méthylation de l'ADN. En effet, l'ADN méthylé peut fixer des protéines appelées Methyl-CpG-binding domain proteins (Meehan et al., 1989). Par la suite, ces protéines peuvent recruter d'autres protéines, telles que les HDACs, provoquant ainsi une compaction de la chromatine (Nan et al., 1998).
5. Utilisation d'inhibiteurs d'HDACs ou de DNMTs
5.1. Les inhibiteurs d'HDACs
14Le rôle répresseur transcriptionnel des HDACs peut être supprimé par certaines molécules appelées inhibiteurs de désacétylases (Marks et al., 2000). L'utilisation de tels inhibiteurs fait partie des nouveaux moyens thérapeutiques envisagés pour lutter contre de nombreuses maladies associées à une dérégulation transcriptionnelle. Ces molécules constituent actuellement un grand espoir de remèdes contre certains cancers. En inhibant les HDACs de classe I et II, ces inhibiteurs provoquent l'arrêt de la prolifération des cellules cancéreuses, l'induction de leur différenciation et/ou leur mise en apoptose (Marks et al., 2001 ; Kramer et al., 2001 ; Kelly et al., 2002). Les inhibiteurs de HDACs peuvent être classés en plusieurs catégories en fonction de leur structure :
15Les hydroxamates. Parmi cette classe, on retrouve notamment la trichostatine A (TSA) et le suberoylanilide hydroxamic acid (SAHA) qui sont deux des inhibiteurs d'HDACs les plus connus. Différentes études ont démontré l'importance de ces deux molécules. Récemment, il a été montré que la TSA inhibait la croissance de lignées de cancer pulmonaire en stoppant le cycle cellulaire et en provoquant l'apoptose via l'activation de la voie des recepteurs de mort et de la voie mitochondriale (Kim et al., 2006 ; Platta et al., 2007). Cependant, la TSA étant très toxique, il est difficile de réaliser des études in vivo. Un essai clinique a récemment démontré l'activité anticancéreuse du SAHA sur des patients atteints d'un mésothéliome, un cancer de la plèvre provoqué par l'inhalation de fibres d'amiante (Krug et al., 2006). Le CBHA (carboxycinnamic acid bishydroxamide) ainsi que ses dérivés (LAQ-824 et sulfonamide hydroxamic acid) possèdent également une activité inhibitrice de HDACs. En effet, le LAQ-824, en combinaison avec l'acide cis-rétinoïque, induit l'apoptose et l'arrêt du cycle cellulaire en phase G2 dans des lignées cellulaires de mélanome en réactivant notamment l'expression de p21 et de RAR β2 (Kato et al., 2007a). Ces inhibiteurs sont généralement actifs à des concentrations de l'ordre d'une centaine de nanomolaires.
16Les peptides cycliques. Ils constituent la classe la plus complexe au niveau structurel. On y retrouve notamment le depsipeptide, la trapoxine et l'apidicine. Ces inhibiteurs sont actifs à des concentrations de l'ordre du nanomolaire. A part le depsipeptide, leur efficacité dans les traitements médicaux reste à prouver. C'est pour cette raison que des analogues du depsipeptide ont été récemment synthétisés. Le depsipeptide induit un arrêt du cycle cellulaire et de l'apoptose associé à une hyperacétylation de l'histone H3 ainsi qu'une augmentation de l'expression de p21 dans des lignées de cancer pulmonaire (Radhakrishnan et al., 2008).
17Les acides aliphatiques. Il s'agit d'une classe comprenant de petits acides gras. Les principaux inhibiteurs de cette classe sont le valproate, le phénylbutyrate et le butyrate de sodium. Ils sont actifs seulement à des concentrations de l'ordre du millimolaire. Différentes études ont montré l'efficacité du valproate dans plusieurs types de cancer comme la leucémie induite par le virus BLV chez le mouton (Achachi et al., 2005), la leucémie promyélocytique aiguë (McMullin et al., 2005) et la leucémie myéloblastique aiguë (Trus et al., 2005). Le valproate permet également une augmentation transitoire de la charge virale chez des patients infectés par le virus HTLV-1 et atteints de HAM/TSP (HTLV-I-Associated Myelopathy/Tropical Spastic Paraparesis) exposant ainsi les cellules infectées à la réponse immunitaire de l'hôte (Lezin et al., 2007). Le butyrate de sodium est, parmi les inhibiteurs d'HDACs, celui qui a été le plus utilisé lors d'études in vitro (Davie, 2003) mais, in vivo, ce composé a été abandonné à cause de sa courte durée de demi-vie.
18Les benzamides. Leur pouvoir inhibiteur d'HDACs est moins puissant que pour les autres classes. La molécule la plus utilisée de cette classe est le MS-275 qui possède des propriétés anticancéreuses intéressantes notamment dans les cancers de la prostate (Qian et al., 2007), des reins (Kato et al., 2007b) et certaines leucémies (Shankar et al., 2005).
19Les cétones électrophiles. Il s'agit d'une classe d'inhibiteurs de désacétylases plus récente incluant la trifluorométhylcétone et l'α-cétoamide. Ils sont actifs à des concentrations proches du micromolaire et sont encore mal connus.
5.2. Les inhibiteurs de DNMTs
20Dans de nombreux cancers, il a été démontré que certains gènes suppresseurs de tumeurs, comme par exemple p16, étaient hyperméthylés (Herman et al., 1995). La conséquence de cette hyperméthylation est l'inactivation de ce gène régulateur du cycle cellulaire. Par contre, les inhibiteurs bloquant l'activité DNA méthyltransférase provoquent l'hypométhylation de ces gènes suppresseurs de tumeurs et possèdent des propriétés anticancéreuses (Santini et al., 2001). Les inhibiteurs de DNMT peuvent être classés en deux groupes : les inhibiteurs nucléosidiques et les inhibiteurs non-nucléosidiques (Lyko et al., 2005).
21Les inhibiteurs nucléosidiques. Ce premier groupe comporte des molécules comme la 5-azacytidine (5-aza-CR), la 5-aza-2'-déoxycytidine (5-aza-CdR ou décitabine) et la zébularine. Ces trois composés fonctionnent comme des analogues de nucléosides et peuvent donc s'insérer dans l'ADN en lieu et place de la cytosine mais ne peuvent pas être méthylés. Toute méthylation de l'ADN néosynthétisé sera donc inhibée après division de la cellule (Jones et al., 1983 ; Worm et al., 2002). La 5-aza-CR et la 5-aza-CdR sont les deux inhibiteurs nucléosidiques les plus utilisés. La 5-aza-CR induit l'apoptose dans des lignées de cancer du colon (Schneider-Stock et al., 2005). L'efficacité de la 5-aza-CdR a été évaluée seule ou en combinaison notamment avec le valproate pour le traitement de la leucémie myéloïde aigüe (Garcia-Manero et al., 2006 ; Blum et al., 2007 ; Soriano et al., 2007). Cependant, ces inhibiteurs de méthylation montrent une forte toxicité et une faible stabilité. Par contre, la zébularine présente une meilleure stabilité (Zhou et al., 2002 ; Cheng et al., 2003) et une plus faible toxicité (Cheng et al., 2004). Il a par ailleurs été démontré que la zébularine, en combinaison avec le SAHA, permettait l'apoptose et l'arrêt du cycle cellulaire de lignées cellulaires de cancer du pancréas (Neureiter et al., 2007).
22Les inhibiteurs non-nucléosidiques. Ce deuxième groupe comprend la procaïne, l'(-)-epigallocatechin-3-O-gallate (EGCG) et le RG108. Ces molécules inhibent les DNMT soit en bloquant le site actif de l'enzyme (EGCG et RG108) soit en se fixant sur les régions CpG de l'ADN empêchant ainsi la liaison des DNMT à l'ADN (procaïne). Ces inhibiteurs sont moins toxiques que les inhibiteurs nucléosidiques. EGCG, un polyphénol présent dans le thé vert, affecte plusieurs voies biologiques (Moyers et al., 2004) et inhibe l'activité méthyltransférase de nombreuses lignées cancéreuses humaines (Fang et al., 2003). Le RG108 bloque l'activité méthyltransférase in vitro, déméthyle l'ADN génomique à des concentrations de l'ordre du micromolaire sans toxicité importante pour les cellules et conduit à la réactivation de certains gènes suppresseurs de tumeurs (Brueckner et al., 2005). La procaïne est également un inhibiteur de DNA méthyltransférase efficace notamment dans des lignées de cancer du sein (Villar-Garea et al., 2003).
5.3. Utilisation de combinaisons d'inhibiteurs
23Bien que les inhibiteurs d'HDACs ou de DNMTs présentent des propriétés anticancéreuses, une des stratégies actuelle consiste à réaliser des combinaisons d'inhibiteurs d'HDACs et de méthylation dans le but d'activer l'expression des gènes anormalement silencieux dans les cellules cancéreuses. Ainsi, une étude clinique récente vient de démontrer l'efficacité d'une combinaison de 5-azacytidine et de phénylbutyrate dans le cadre de tumeurs solides ainsi que de leucémies (Rudek et al., 2005). D'autre part, l'utilisation d'une combinaison de valproate et de décitabine a révélé un effet antileucémique en culture cellulaire (Yang et al., 2005). Par ailleurs, les inhibiteurs d'HDACs et de méthylation de l'ADN peuvent également être associés à des composés classiquement utilisés lors de chimiothérapies. L'avantage de cette approche est tout d'abord de se baser sur un traitement existant partiellement efficace. Ensuite, le mélange de molécules possédant des modes d'action très différents peut engendrer un effet de synergie dirigé contre les cellules cancéreuses. Enfin, cette approche pourrait permettre de réduire les concentrations requises du composé chimiothérapeutique, limitant de cette manière les effets secondaires. C'est ainsi que l'utilisation du LAQ-824 permet de sensibiliser les cellules de cancer du sein au taxotère et à la gemcitabine (Fuino et al., 2003). Le phénylbutyrate sensibilise des cellules B tumorales à l'action de différentes molécules comme la doxorubicine, l'étoposide, le cisplatine ou encore la fludarabine (Witzig et al., 2000). De même, le depsipeptide augmente l'effet thérapeutique du docétaxel, un agent chimiothérapeutique utilisé notamment dans le traitement du cancer de la prostate (Zhang et al., 2007). Il est également possible de créer de nouvelles molécules possédant à la fois une extrémité inhibitrice d'HDACs et une extrémité inhibitrice de DNMTs. Ces molécules hybrides (appelées Twin drugs) sont actuellement testées dans notre laboratoire sur des moutons leucémiques infectés par le virus de la leucémie bovine (BLV) et sur des lignées de mésothéliome pleural.
6. Conclusion
24Les cancers sont l'une des causes principales de mortalité dans le monde. Dans la plupart des cas, les taux de réponse aux agents utilisés en chimiothérapie sont assez faibles et avoisinent les 20 %. Les modifications épigénétiques constituent un mécanisme important dans la régulation de l'expression des gènes. Au cours de l'évolution vers le cancer, les processus épigénétiques sont perturbés notamment au niveau de la méthylation de l'ADN ou de l'acétylation des histones. Des inhibiteurs de ces mécanismes constituent une classe grandissante de composés anticancéreux.
25Remerciements
26Nous remercions le Fonds national de la Recherche scientifique (FNRS) ainsi que l'opération Télévie pour leur support financier. Fabian Vandermeers (boursier Télévie), Richard Kettmann (Directeur de Recherches) et Luc Willems (Directeur de Recherches) sont membres du FNRS.
Bibliographie
Achachi A. et al., 2005. Valproate activates bovine leukemia virus gene expression, triggers apoptosis, and induces leukemia/lymphoma regression in vivo. Proc. Natl Acad. Sci. U.S.A, 102, 10309-10314.
Ahmad K. & Henikoff S., 2002. Histone H3 variants specify modes of chromatin assembly. Proc. Natl Acad. Sci. U.S.A, 99(Suppl. 4), 16477-16484.
Berger S.L., 2002. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev., 12, 142-148.
Bird A., 2002. DNA methylation patterns and epigenetic memory. Genes Dev., 16, 6-21.
Blum W. et al., 2007. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J. Clin. Oncol., 25, 3884-3891.
Brueckner B. et al., 2005. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res., 65, 6305-6311.
Cheng J.C. et al., 2003. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl Cancer Inst., 95, 399-409.
Cheng J.C. et al., 2004. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol. Cell Biol., 24, 1270-1278.
Cheung P. & Lau P., 2005. Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol., 19, 563-573.
Cuthbert G.L. et al., 2004. Histone deimination antagonizes arginine methylation. Cell, 118, 545-553.
Davie J.R., 2003. Inhibition of histone deacetylase activity by butyrate. J. Nutr., 133, 2485S-2493S.
de Ruijter A.J. et al., 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J., 370, 737-749.
Drummond D.C. et al., 2005. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol., 45, 495-528.
Fang M.Z. et al., 2003. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res., 63, 7563-7570.
Felsenfeld G. & Groudine M., 2003. Controlling the double helix. Nature, 421, 448-453.
Fuino L. et al., 2003. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol. Cancer Ther., 2, 971-984.
Garcia-Manero G. et al., 2006. Phase 1/2 study of the combination of 5-aza-2'-deoxycytidine with valproic acid in patients with leukemia. Blood, 108, 3271-3279.
Grabher A. et al., 1994. Subcellular location of enzymes involved in core histone acetylation. Biochemistry, 33, 14887-14895.
Gray S.G. & Ekstrom T.J., 2001. The human histone deacetylase family. Exp. Cell Res., 262, 75-83.
Herman J.G. et al., 1995. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res., 55, 4525-4530.
Humphrey G.W. et al., 2001. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J. Biol. Chem., 276, 6817-6824.
Jason L.J. et al., 2002. Histone ubiquitination: a tagging tail unfolds? Bioessays, 24, 166-174.
Jones P.A., Taylor S.M. & Wilson V.L., 1983. Inhibition of DNA methylation by 5-azacytidine. Recent Results Cancer Res., 84, 202-211.
Kato Y. et al., 2007a. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol. Cancer Ther., 6, 70-81.
Kato Y. et al., 2007b. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin. Cancer Res., 13, 4538-4546.
Kelly W.K., O'Connor O.A. & Marks P.A., 2002. Histone deacetylase inhibitors: from target to clinical trials. Expert Opin. Invest. Drugs, 11, 1695-1713.
Kim H.R. et al., 2006. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp. Mol. Med., 38, 616-624.
Kornberg R.D., 1974. Chromatin structure: a repeating unit of histones and DNA. Science, 184, 868-871.
Kornberg R.D. & Lorch Y., 1995. Interplay between chromatin structure and transcription. Curr. Opin. Cell Biol., 7, 371-375.
Kramer O.H., Gottlicher M. & Heinzel T., 2001. Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab., 12, 294-300.
Krug L.M. et al., 2006. Potential role of histone deacetylase inhibitors in mesothelioma: clinical experience with suberoylanilide hydroxamic acid. Clin. Lung Cancer, 7, 257-261.
Kuo M.H. & Allis C.D., 1998. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays, 20, 615-626.
Lacoste N. & Cote J., 2003. The epigenetic code of histones. Med. Sci., 19, 955-959.
Leonhardt H., Page A.W., Weier H.U. & Bestor T.H., 1992. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell, 71, 865-873.
Lezin A. et al., 2007. Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1-associated myelopathy/tropical spastic paraparesis patients. Blood, 110, 3722-3728.
Liu Y., Oakeley E.J., Sun L. & Jost J.P., 1998. Multiple domains are involved in the targeting of the mouse DNA methyltransferase to the DNA replication foci. Nucleic Acids Res., 26, 1038-1045.
Luger K. et al., 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature, 389, 251-260.
Lyko F. & Brown R., 2005. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl Cancer Inst., 97, 1498-1506.
Marks P.A., Richon V.M., Breslow R. & Rifkind R.A., 2001. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol., 13, 477-483.
Marks P.A., Richon V.M. & Rifkind R.A., 2000. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl Cancer Inst., 92, 1210-1216.
McMullin M.F. et al., 2005. Prolonged molecular remission in PML-RARalpha-positive acute promyelocytic leukemia treated with minimal chemotherapy followed by maintenance including the histone deacetylase inhibitor sodium valproate. Leukemia, 19, 1676-1677.
Meehan R.R. et al., 1989. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell, 58, 499-507.
Moyers S.B. & Kumar N.B., 2004. Green tea polyphenols and cancer chemoprevention: multiple mechanisms and endpoints for phase II trials. Nutr. Rev., 62, 204-211.
Nan X. et al., 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393, 386-389.
Neureiter D. et al., 2007. Apoptosis, proliferation and differentiation patterns are influenced by Zebularine and SAHA in pancreatic cancer models. Scand. J. Gastroenterol., 42, 103-116.
Okano M., Bell D.W., Haber D.A. & Li E., 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99, 247-257.
Platta C.S., Greenblatt D.Y., Kunnimalaiyaan M. & Chen H., 2007. The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. J. Surg. Res., 142, 219-226.
Qian D.Z. et al., 2007. Antitumor activity of the histone deacetylase inhibitor MS-275 in prostate cancer models. Prostate, 67, 1182-1193.
Radhakrishnan V., Song Y.S. & Thiruvengadam D., 2008. Romidepsin (depsipeptide) induced cell cycle arrest, apoptosis and histone hyperacetylation in lung carcinoma cells (A549) are associated with increase in p21 and hypophosphorylated retinoblastoma proteins expression. Biomed. Pharmacother., 62(2), 85-93.
Redon C. et al., 2002. Histone H2A variants H2AX and H2AZ. Curr. Opin. Genet. Dev., 12, 162-169.
Rudek M.A. et al., 2005. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J. Clin. Oncol., 23, 3906-3911.
Santini V., Kantarjian H.M. & Issa J.P., 2001. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann. Intern. Med., 134, 573-586.
Schneider-Stock R. et al., 2005. 5-Aza-cytidine is a potent inhibitor of DNA methyltransferase 3a and induces apoptosis in HCT-116 colon cancer cells via Gadd45- and p53-dependent mechanisms. J. Pharmacol. Exp. Ther., 312, 525-536.
Shankar S. et al., 2005. Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: involvement of both death receptor and mitochondrial pathways. Int. J. Mol. Med., 16, 1125-1138.
Soriano A.O. et al., 2007. Safety and clinical activity of the combination of 5-azacytidine, valproic acid and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood, 110, 2302-2308.
Strahl B.D. & Allis C.D., 2000. The language of covalent histone modifications. Nature, 403, 41-45.
Trus M.R. et al., 2005. The histone deacetylase inhibitor valproic acid alters sensitivity towards all trans retinoic acid in acute myeloblastic leukemia cells. Leukemia, 19, 1161-1168.
Uberbacher E.C. & Bunick G.J., 1985. X-ray structure of the nucleosome core particle. J. Biomol. Struct. Dyn., 2, 1033-1055.
Villar-Garea A., Fraga M.F., Espada J. & Esteller M., 2003. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res., 63, 4984-4989.
Wade P.A., 2001. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum. Mol. Genet., 10, 693-698.
Watson J.D. & Crick F.H., 1953. The structure of DNA. Cold Spring Harb. Symp. Quant. Biol., 18, 123-131.
Witzig T.E., 2000. Induction of apoptosis in malignant B cells by phenylbutyrate or phenylacetate in combination with chemotherapeutic agents. Clin. Cancer Res., 6, 681-692.
Worm J. & Guldberg P., 2002. DNA methylation: an epigenetic pathway to cancer and a promising target for anticancer therapy. J. Oral Pathol. Med., 31, 443-449.
Yang H. et al., 2005. Antileukemia activity of the combination of 5-aza-2'-deoxycytidine with valproic acid. Leuk. Res., 29, 739-748.
Yoder J.A., Soman N.S., Verdine G.L. & Bestor T.H., 1997. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol., 270, 385-395.
Zhang Y. & Reinberg D., 2001. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15, 2343-2360.
Zhang Z. et al., 2007. Enhanced therapeutic effect on androgen-independent prostate cancer by depsipeptide (FK228), a histone deacetylase inhibitor, in combination with docetaxel. Urology, 70, 396-401.
Zhou L. et al., 2002. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J. Mol. Biol., 321, 591-599.
Pour citer cet article
A propos de : Fabian Vandermeers
Gembloux Agricultural University – FUSAGx. Unité de Biologie cellulaire et moléculaire. Avenue Maréchal Juin, 13. B-5030 Gembloux (Belgique). E-mail : vandermeers.f@fsagx.ac.be
A propos de : Richard Kettmann
Gembloux Agricultural University – FUSAGx. Unité de Biologie cellulaire et moléculaire. Avenue Maréchal Juin, 13. B-5030 Gembloux (Belgique).
A propos de : Luc Willems
Gembloux Agricultural University – FUSAGx. Unité de Biologie cellulaire et moléculaire. Avenue Maréchal Juin, 13. B-5030 Gembloux (Belgique).
